Autor: Josu Galarza
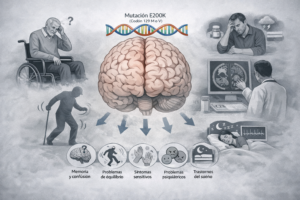
La enfermedad de Huntington es un trastorno neurodegenerativo clasificado como enfermedad rara, con una incidencia de entre 5 y 10 casos por cada 100.000 habitantes. De carácter hereditario, sus principales manifestaciones clínicas son los movimientos involuntarios, así como alteraciones cognitivas y psiquiátricas. La presencia de agregados proteicos tóxicos en el cerebro, junto a su mecanismo de propagación, ha llevado a considerarla una enfermedad de tipo priónico.
Un reciente estudio —cuyos datos aún no han sido publicados en su totalidad— ha demostrado la capacidad para ralentizar la progresión de la enfermedad, mejorando la autonomía y el bienestar de los pacientes. El estudio, comunicado mediante una nota de prensa por la empresa neerlandesa uniQure (nota de prensa original en inglés aquí), emplea la terapia génica como herramienta para silenciar el gen responsable de la toxicidad cerebral de los pacientes con Huntington, retrasando así su deterioro cognitivo y motor de los pacientes. Este punto es de vital importancia, ya que el curso clínico de esta enfermedad puede prolongarse entre 15 y 20 años, durante los cuales los pacientes soportan fuertes dolores musculares debido a la aparición de movimientos involuntarios persistentes.
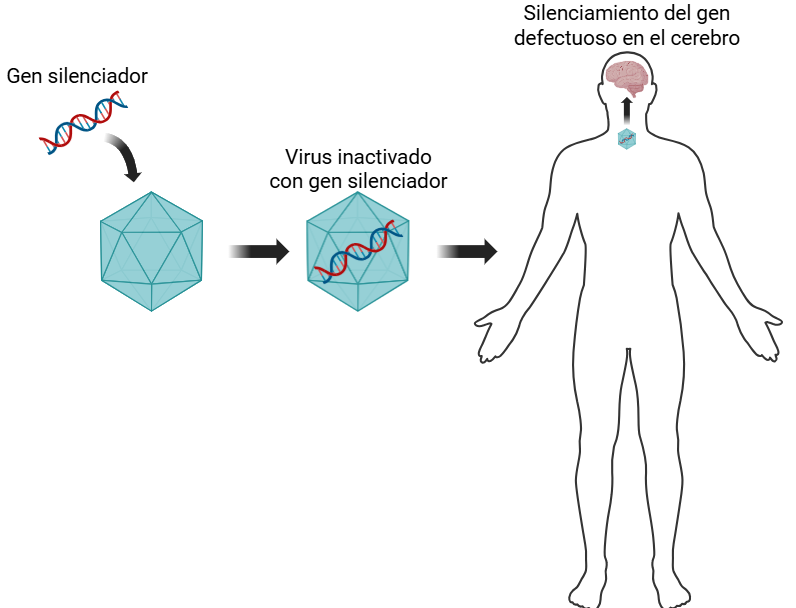
El fármaco, denominado AMT-130, está compuesto por un tipo de virus inofensivo llamado AAV (virus adenoasociado) capaz de introducir un fragmento de ADN en las células. Esta secuencia de ADN está diseñada para unirse al gen afectado e impedir la producción de la proteína tóxica, frenando así el avance de la enfermedad. La administración del tratamiento se realiza mediante infusión directa en el sistema nervioso central a través de catéteres en las regiones cerebrales más afectadas. Los resultados son prometedores: los 12 pacientes que recibieron la dosis más alta del fármaco fueron observados durante 3 años, y mostraron una reducción media de 0.38 puntos en la escala de evaluación de la enfermedad de Huntington, frente a una reducción de 1.52 puntos en el grupo de comparación, lo que representa una mejora del 75%. También mostraron menor deterioro en otras pruebas clínicas y una reducción del 8.2% en una proteína del líquido cefalorraquídeo llamada neurofilamento ligero, un marcador que refleja la muerte neuronal.
Tras concluir esta fase I/II del estudio, uniQure pretende lograr la aprobación para su comercialización a lo largo del próximo año. El visto bueno de las autoridades sanitarias y los buenos resultados de este estudio son determinantes para la aceptación de las futuras terapias génicas que empleen estrategias similares, abriendo las puertas al estudio de terapias para otras enfermedades, incluyendo las enfermedades priónicas.