Author: Cristina Sampedro
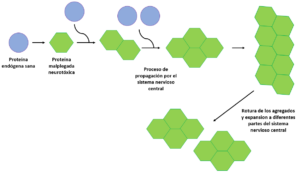
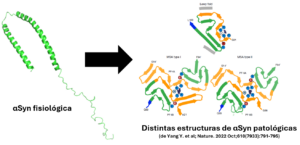
Prion diseases can occur spontaneously or genetically, the latter being caused by a mutation in the prion protein (PrP); these diseases are rare but devastating neurodegenerative disorders that affect both humans and animals. The main types of genetic prion diseases are genetic Creutzfeldt-Jakob disease (gCJD), fatal familial insomnia (FFI) and Gerstmann-Sträussler-Scheinker syndrome (GSS). Among these, the most common is gCJD caused by the E200K mutation in the prion protein (PrP). In simple terms, this mutation causes the 200th position of the protein to change from the amino acid glutamate to a lysine. This and other mutations are believed to cause disease by making PrP more susceptible to misfolding or conversion to its pathogenic form (PrPSc).
A recently published discovery by researchers at Boston University has revealed that the architecture of the regions of contact between neurons, known as synapses, is altered in neurons that produce PrP with the E200K mutation even in the absence of the pathogenic form. In other words, this means that alterations are seen at these synapses before damage from the misfolded protein is seen, indicating that there may be toxicity due to the absence of healthy cellular PrP, and not just due to the deleterious effect of PrP.Sc.
The study, published in the journal Stem Cell Reports, opens a new window of opportunity by suggesting that there may be detectable defects in neurons long before the first symptoms of an inherited prion disease appear.
To carry out this study, the researchers used an innovative technology to generate induced pluripotent stem cells. To do this, they used blood cells from a family in which some individuals carried the E200K mutation and reverted them to a state similar to that of embryonic cells. These special cells, which due to their resemblance to embryonic cells have the capacity to generate all the different cell types that make up the organism, can be transformed in the laboratory in a targeted way into the cells of interest to the researcher. In this case, these cells of carriers and non-carriers of the E200K mutation belonging to the same family were transformed into neurons in order to study the effect of this mutation on them.
To ensure the accuracy of their findings, the researchers were able to compare the effect of this mutation in neurons with a very similar genetic background, as they worked with samples from individuals from the same family. This helps to confirm that the defects seen in the neurons affected by the mutation are due to that mutation and not to other genetic factors that the donors may have. In addition, to obtain even more conclusive results, they used the novel CRISPR/Cas9 technology, a high-precision gene-editing tool that allows the DNA of cells to be edited on the spot to correct the mutation in neurons from carriers of the mutation. In this way, neuronal cultures that have exactly the same genetic background, except for the E200K mutation, can be compared.
Although the study does not conclude whether the observed effect is due to the mutated protein acquiring a toxic function or to the loss of function of the cellular protein, previous research in animals suggests that the former is more likely, as there appears to be no developmental effect on neurons in animals that do not express cellular PrP.
Looking ahead, the researchers believe that the use of induced pluripotent stem cell cultures represents a significant advance towards personalised medicine, as they can become a powerful platform for investigating and testing future therapies using cells from the patients themselves who would receive the therapy. This approach would not only allow the development of more specific treatments, but also the design of therapies targeted to the specific effects that different mutations have on the state of neurons.
Link to original article (in English).