Auteur: Josu Galarza
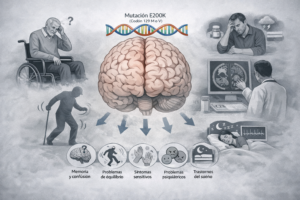
La maladie de Huntington est une affection neurodégénérative classée parmi les maladies rares, avec une incidence comprise entre 5 et 10 cas pour 100 000 habitants. De nature héréditaire, ses principales manifestations cliniques sont des mouvements involontaires, ainsi que des troubles cognitifs et psychiatriques. La présence d'agrégats protéiques toxiques dans le cerveau, associée à son mécanisme de propagation, a conduit à la considérer comme une maladie de type prionique.
Une étude récente, dont les données n'ont pas encore été publiées dans leur intégralité, a démontré la capacité à ralentir la progression de la maladie, améliorant ainsi l'autonomie et le bien-être des patients. Cette étude, communiquée par un communiqué de presse de la société néerlandaise uniQure (communiqué de presse original en anglais ici) , utilise la thérapie génique comme outil pour désactiver le gène responsable de la toxicité cérébrale des patients atteints de la maladie de Huntington, retardant ainsi leur détérioration cognitive et motrice. Ce point est d'une importance vitale, car l'évolution clinique de cette maladie peut s'étendre sur 15 à 20 ans, pendant lesquels les patients souffrent de fortes douleurs musculaires dues à l'apparition de mouvements involontaires persistants.
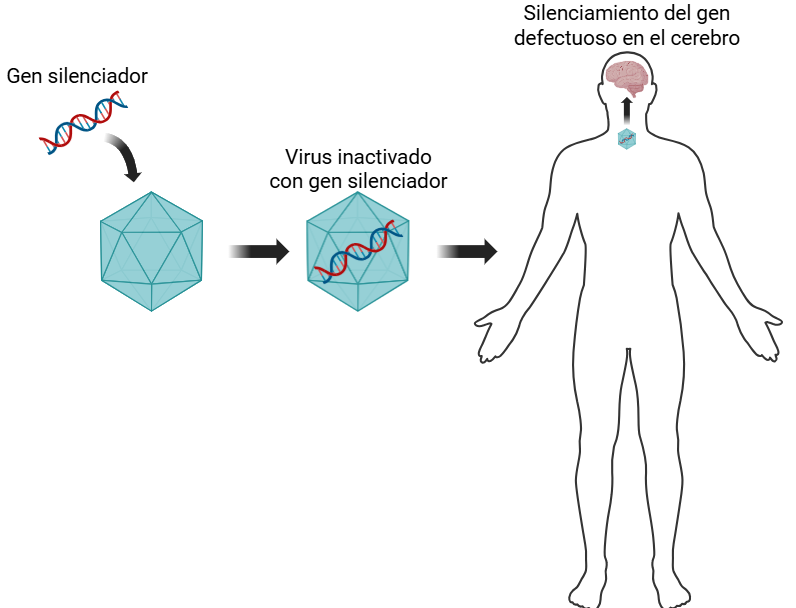
Le médicament, appelé AMT-130, est composé d'un type de virus inoffensif appelé AAV (virus adéno-associé) capable d'introduire un fragment d'ADN dans les cellules. Cette séquence d'ADN est conçue pour se lier au gène affecté et empêcher la production de la protéine toxique, freinant ainsi la progression de la maladie. Le traitement est administré par perfusion directe dans le système nerveux central à l'aide de cathéters placés dans les régions cérébrales les plus touchées. Les résultats sont prometteurs : les 12 patients ayant reçu la dose la plus élevée du médicament ont été suivis pendant 3 ans et ont montré une réduction moyenne de 0,38 point sur l'échelle d'évaluation de la maladie de Huntington, contre une réduction de 1,52 point dans le groupe témoin, soit une amélioration de 75 %. Ils ont également montré une moindre détérioration dans d'autres tests cliniques et une réduction de 8,2 % d'une protéine du liquide céphalo-rachidien appelée neurofilament léger, un marqueur qui reflète la mort neuronale.
Une fois cette phase I/II de l'étude terminée, uniQure prévoit d'obtenir l'autorisation de commercialisation au cours de l'année prochaine. L'accord des autorités sanitaires et les bons résultats de cette étude sont déterminants pour l'acceptation des futures thérapies géniques utilisant des stratégies similaires, ouvrant ainsi la voie à l'étude de thérapies pour d'autres maladies, y compris les maladies à prions.