Autor: Hasier Eraña
Desde el laboratorio de Sonia Vallabh y Eric Minikel, en el Broad Institute de Boston, llegan novedades de las que informa el propio Eric en su blog (cureffi.org). El pasado 14 de abril, estos investigadores consiguieron un hito poco común en la investigación académica (aquella que se hace fuera de la industria farmacéutica), la autorización de un Nuevo Medicamento en Investigación (IND, por sus siglas en inglés: Investigational New Drug) por parte de la FDA (Food and Drug Administration de EE.UU., agencia responsable de la autorización de ensayos en humanos de nuevos fármacos). Este hecho, supone un importante paso adelante en la larga y costosa vía que lleva del descubrimiento al desarrollo y comercialización de una nueva terapia que, en este caso, trata de una novedosa aproximación para reducir los niveles de PrPC en el cerebro utilizando siRNA divalente. Aunque los propios investigadores reconocen que aún queda un largo camino por recorrer para probar la eficacia y seguridad de este nuevo tratamiento en pacientes con enfermedades priónicas, a través de una completa publicación en su blog, han querido celebrar la buena noticia, detallando el estado actual de desarrollo de esta potencial terapia, y describiendo los siguientes pasos del proceso. A continuación, resumo el contenido del artículo publicado por Eric Minikel el pasado 14 de abril, cuya versión original puede verse en cureffi.org.
El viaje hacia un fármaco candidato
En 2019, la Dra. Anastasia Khvorova de la Escuela de Medicina de la Universidad de Massachussets visitó el Broad Institute para hablar sobre su trayectoria profesional en la ingeniería de mejores fármacos de ARN de interferencia corto o siRNA. Se centró en un hito que entonces era muy reciente: la invención por parte de su laboratorio de lo que ellos llaman siRNA divalente.
Hasta ese momento, los oligonucleótidos antisentido (ASO) eran la única opción para reducir una proteína diana en el cerebro (Ahora están en un ensayo de Fase I gracias a la colaboración con Ionis Pharmaceuticals). Pero necesitábamos otra oportunidad de gol. La publicación sobre el siRNA divalente de Anastasia mostró una reducción muy profunda y duradera de un gen diana (HTT), y pensamos que, si podíamos hacer esto con nuestro gen, PRNP, sería realmente prometedor.
Su equipo examinó rápidamente secuencias de siRNA contra los genes de ratón y humano, y presentó una patente sobre varias de ellas [WO2021173984]. Combinando los datos preliminares de su laboratorio sobre secuencias de siRNA y los datos preliminares de nuestro laboratorio sobre nuevos modelos de ratón con PrP humano y un ensayo para cuantificar la reducción de PrP en el cerebro, solicitamos una subvención de los Institutos Nacionales de Salud (NIH) para apoyar el desarrollo de un fármaco. Sorprendentemente, la obtuvimos.
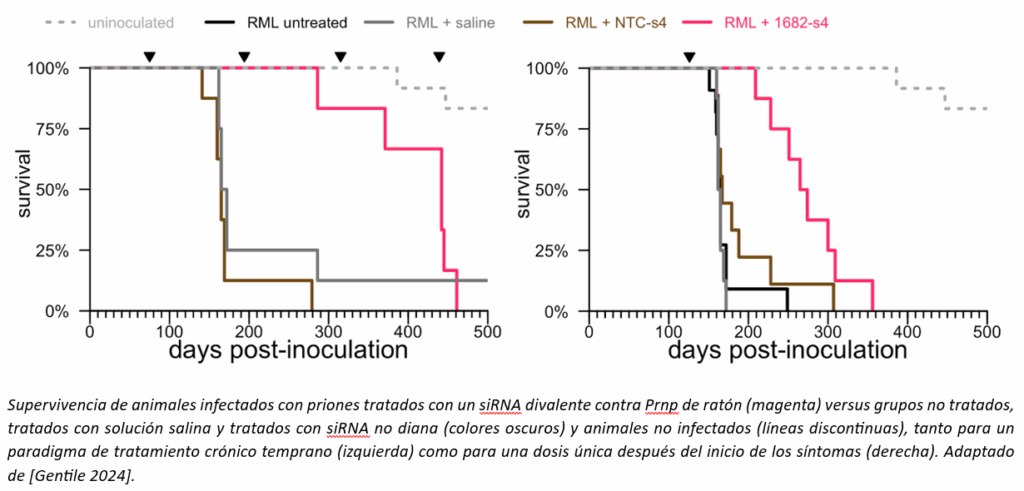
Probamos las moléculas de siRNA enviadas desde la Universidad de Massachusetts, primero en células y luego en ratones, para encontrar la mejor molécula. El mejor siRNA que pudimos encontrar contra Prnp de ratón no era excepcionalmente potente; solo redujo la PrP en todo el cerebro del ratón en aproximadamente un 50%. Pero eso fue suficiente como para preguntar: ¿la reducción de PrP con un siRNA divalente mejora la supervivencia, tal como habíamos visto con los ASO? Estos son experimentos de larga duración, donde un grupo de animales debe ser infectado con priones y, meses después, recibir el fármaco en un momento muy específico. El experimento funcionó: el tratamiento con siRNA divalente, incluso una sola dosis justo después del inicio de los síntomas, marcó una gran diferencia en la vida útil de los animales infectados con priones.
Mientras tanto, para el otoño de 2022, nos habíamos decidido por un único compuesto de siRNA divalente contra el PRNP humano que, según todos los experimentos que habíamos podido realizar, era el mejor del grupo. Vimos una profundidad de reducción de PrP que nunca habíamos visto antes, con meses de durabilidad, y parecía ser bien tolerado. Deseábamos desesperadamente que este fuera otro caballo en la carrera para tratar la enfermedad priónica. Las opciones eran claras: o nos dedicábamos a desarrollar este fármaco nosotros mismos, o aceptábamos la cruda realidad de que todo este trabajo sería «solo un estudio en ratones», otro artículo científico más que dice «esto parece prometedor» pero que luego nunca llega a la vida de ningún ser humano.
Casi al mismo tiempo, nos enteramos de un nuevo mecanismo de financiación del NIH, un programa enfocado precisamente en nuestra situación: tienes datos prometedores en ratones, pero una primera dosis en humanos está a años de trabajo y millones de dólares de distancia. Este programa parecía ser nuestra única oportunidad, pero no había duda: incluso con el apoyo financiero del NIH para hacer la tarea factible, esto iba a ser un esfuerzo muy, muy grande para nosotros.
Un paréntesis científico: ¿Cómo funciona el siRNA divalente?
siRNA significa ARN de interferencia corto. Este tipo de fármaco está compuesto por los mismos bloques de construcción que el ADN y el ARN (A, C, G y T o U) y actúa a través del mecanismo de interferencia de ARN (RNAi). El RNAi se descubrió en 1998 y les valió a sus descubridores el Premio Nobel de Fisiología o Medicina en 2006. El fármaco siRNA está diseñado para unirse de forma específica a una molécula de ARN diana en la célula, en este caso la que da lugar a la PrP. El resultado es menos ARN diana y, por lo tanto, menos de la proteína que codifica. Por lo tanto, un siRNA es una forma de reducir la cantidad de una proteína específica causante de enfermedades. El primer fármaco siRNA, Patisirán para la amiloidosis por transtiretina, recibió la aprobación de la FDA en 2018.
Un siRNA es similar a un ASO en que es un fármaco diseñado para unirse a un ARN diana y degradarlo. Al igual que un ASO, en humanos este siRNA se administraría en el líquido cefalorraquídeo (LCR) mediante una inyección intratecal, en otras palabras, una punción lumbar (PL). Un siRNA es diferente de un ASO en que es un oligonucleótido de doble cadena (en contraposición a una sola cadena), utiliza diferentes patrones de modificación química para estabilizarlo y actúa a través de una vía celular diferente (RISC en contraposición a la ARNasa H1).
Hasta el año pasado, había 6 fármacos siRNA aprobados por la FDA, y decenas más en ensayos clínicos. Varias compañías farmacéuticas diferentes están desarrollando siRNA con: diferentes modificaciones químicas, diferentes conjugados para promover la captación en diferentes tejidos del cuerpo y diferentes secuencias para diferentes dianas en diferentes indicaciones de enfermedades.
Un siRNA divalente es un tipo especial desarrollado por el laboratorio de Anastasia Khvorova. Consiste en dos copias del mismo siRNA de doble cadena, unidas entre sí. Empíricamente, estudios en ratones realizados por el laboratorio de Khvorova cuando estaban desarrollando esta tecnología encontraron que los siRNA simples (monovalentes) con su química particular simplemente no eran captados ni retenidos en el cerebro. Cuando unieron dos de ellos para formar una molécula divalente, de repente se retuvo una gran cantidad de fármaco en el cerebro, fue captado por las células y fue farmacológicamente activo.
Escalando la montaña del IND (Nuevo Medicamento en Investigación)
En los EE. UU., y realmente en todos los países de la Tierra, los pasos necesarios para pasar de «se ve bien en un ratón» al permiso para dosificar a una persona no solo requieren mucho tiempo y son costosos, sino que también son estudios altamente especializados. Existe toda una industria que, si no trabajas en el desarrollo de fármacos, nunca has visto ni pensado, toda dirigida por empresas cuyos nombres no conoces. La síntesis química, la purificación, las pruebas de calidad y el envasado del fármaco (Química, Fabricación y Controles, CMC, en la jerga de la industria) deben realizarse bajo las Buenas Prácticas de Fabricación (GMP), que son 10 veces más caras que fabricar un fármaco para utilizar en ratones. Primero, se siguen todos los pasos de GMP para fabricar una sustancia farmacológica (DS), un polvo blanco, luego se sigue un proceso adicional completo para formular ese polvo en un producto farmacológico (DP), que podría ser una píldora o, en nuestro caso, un vial de líquido inyectable estéril. Los estudios de toxicología deben realizarse bajo las Buenas Prácticas de Laboratorio (GLP), con decenas o cientos de animales, cada uno produciendo cientos o incluso miles de mediciones y lecturas para determinar si el fármaco es tóxico. Se analizan la sangre y/o los tejidos de los animales utilizando un ensayo farmacocinético (PK) validado por GLP para confirmar cuánto fármaco hay realmente y asegurarse de que fueron dosificados correctamente. Hay estudios de interacción fármaco-fármaco (DDI), para ver si a los pacientes que toman alguno de los cientos de fármacos aprobados se les debe prohibir tomar su fármaco. Hay estudios de genotoxicidad, para determinar si el fármaco puede mutar el ADN y causar cáncer o defectos de nacimiento.
El objetivo de todo esto es llevarlo a otro acrónimo más: el IND, o Nuevo Fármaco en Investigación. Una solicitud de IND es el paquete de datos de todos los estudios anteriores que el desarrollador o patrocinador de un fármaco presenta a la FDA para solicitar permiso para dosificar a un ser humano con un nuevo fármaco por primera vez.
Desde que presentamos nuestra propuesta al NIH en octubre de 2022, sabíamos que pasaría la mayor parte de un año antes de que se pudiera otorgar la financiación, más probablemente algún tiempo de retraso después de eso antes de que se pudieran establecer los contratos y el trabajo pudiera comenzar en serio. Con el deseo de avanzar lo más rápido posible, iniciamos la fabricación del fármaco de inmediato utilizando fondos de Prion Alliance. En junio de 2023, Sonia y yo tuvimos nuestra primera reunión Pre-IND con la FDA, en la que pudimos hacer preguntas sobre lo que se requeriría para nuestro IND dadas las particularidades de nuestro fármaco y nuestra enfermedad específicos. Con el asesoramiento de la FDA en mano, trabajamos con algunos donantes extremadamente inteligentes, dedicados y apasionados para recaudar fondos para algunos de los otros pasos más limitantes, particularmente la primera ronda de estudios de toxicología y el desarrollo de un ensayo de farmacocinética (PK), el método para cuantificar el fármaco en sangre o tejidos.
Nuestros recursos financieros aún eran bastante limitados, y sentimos que la velocidad era primordial, por lo que hicimos algunas concesiones difíciles. Optamos por realizar un estudio de toxicología de dosis única en animales, lo cual, la FDA nos dejó muy claro, solo nos permitiría realizar un ensayo de dosis única en pacientes. Tomamos esta decisión por varias razones. Solo podíamos permitirnos un estudio de dosis única, y un estudio de dosis única sería más rápido. Nuestros datos en ratones mostraron que una sola dosis podría tener una actividad que duraría de 4 a 6 meses, y una sola dosis en una etapa sintomática de la enfermedad prolongó la supervivencia en ratones infectados con priones en 3,5 meses. Por lo tanto, considerando que muchos pacientes con enfermedad priónica viven solo unos pocos meses desde el diagnóstico, sentimos que incluso una sola dosis tenía el potencial de ser clínicamente relevante. Finalmente, un estudio de toxicología de dosis única presentaba menos riesgo. Por regla general, los estudios de toxicología ya se realizan normalmente con varias veces el equivalente de la dosis que espera usar en humanos. Si a esto se añade una administración mensual durante 9 meses (como es típico en los estudios de toxicidad crónica), los animales reciben una cantidad muy elevada de fármaco, lo que aumenta considerablemente la probabilidad de que aparezcan efectos tóxicos que impidan avanzar hacia ensayos clínicos.
En un increíble golpe de buena fortuna, nuestra propuesta de subvención al NIH finalmente fue financiada. Esto nos dio un camino a seguir para realizar todos los estudios restantes necesarios para obtener un IND. Este proceso requirió 2 años y medio de intenso trabajo, realizado conjuntamente y en gran medida liderado por la Responsable de Proyecto, Alissa Coffey.
A principios de 2025, todos los estudios habilitadores del IND estaban terminados, y lo que quedaba era escribir el IND en sí. A pesar de que teníamos consultores, encontrar plantillas y ejemplos para trabajar fue un desafío importante. La industria realiza sus IND con un estándar industrial, que es incluso más de lo que se requiere para un IND iniciado por un investigador. Los académicos que han presentado IND, y no hay demasiados ejemplos para empezar, generalmente no lo han hecho para modalidades de fármacos novedosas, como lo es el siRNA divalente. Y los IND, ya sean industriales o académicos, que se pueden consultar, son muy difíciles de conseguir. Pero muy generosamente, algunas personas compartieron sus IND conmigo, así que tuve algún tipo de punto de partida para trabajar. El 5 de febrero de 2025, Alissa y yo nos sentamos juntas en mi oficina e hicimos clic en enviar.
Lo que significa nuestro IND abierto
Cuando la FDA autoriza un IND, no solo autoriza un fármaco, sino que autoriza un lote específico de fármaco para una población de pacientes específica, tratados según un protocolo de ensayo clínico específico. Cada detalle debe especificarse. En nuestro IND, presentamos dos protocolos de ensayo: uno para tratar a pacientes sintomáticos ya diagnosticados con enfermedad priónica, y otro para tratar a pacientes presintomáticos en riesgo de enfermedad priónica. El 14 de marzo, la FDA respondió que «podemos proceder» en pacientes sintomáticos, mientras que estamos en «suspensión clínica parcial» para pacientes presintomáticos. Eso significa que quieren ver más datos antes de permitirnos dosificar a personas presintomáticas, y nos han enviado una carta detallada que describe todo lo que necesitaremos hacer para desbloquear la dosificación presintomática.
Pero mientras tanto, tenemos un IND abierto: la FDA nos permite iniciar un ensayo clínico en pacientes sintomáticos con enfermedad priónica. Ojalá eso significara que podríamos empezar a dosificar a la gente mañana, pero la realidad es más complicada. Primero, todavía no tenemos la financiación para llevar a cabo un ensayo clínico. El NIH también tiene un programa para esto, y tras superar con éxito la Etapa 1 para este mecanismo de financiación, y a los pocos días de recibir respuesta de la FDA, presentamos nuestra propuesta de Etapa 2. Si esto se financiará y cuándo, sigue siendo una incógnita. Lo más pronto que podría suceder es dentro de un par de meses. Pero también podría ser varios meses, o nunca.
Supongamos que alguien nos extendiera un cheque hoy por el costo total de realizar un ensayo. Aun así, llevaría tiempo ponerlo en marcha. Un ensayo clínico es una empresa extremadamente compleja. Además, si el NIH financia el ensayo, determinará varios detalles de cómo se llevará a cabo y, una vez obtenida la aprobación ética, aún quedará una larga lista de tareas por completar.
Estoy más ansioso que nadie por conocer el cronograma de cuándo comenzaremos a dosificar a las personas, pero hay grandes incógnitas en este momento, incluido que aún no podemos descartar que la respuesta sea «nunca», si no podemos encontrar la financiación. Cuando he hablado con personas con experiencia en ensayos clínicos “de primera vez en humanos” (conocidos en inglés como “first-in-human trials”), la mayoría me ha dicho que, si la financiación llega pronto, sería posible administrar el fármaco a un primer paciente hacia finales de 2025.
Qué esperar cuando estemos listos para administrar el fármaco a pacientes
Si la financiación lo permite, en algún momento de los próximos meses podremos anunciar que un ensayo clínico está abierto y se empezarían a aceptar a pacientes. Aquí hay algunas cosas que deberá tener en cuenta cuando eso suceda:
Primero, un ensayo clínico es investigación, no tratamiento. Estamos realizando un ensayo clínico precisamente porque aún no sabemos si el fármaco es seguro o eficaz. Todos los fármacos causan algún tipo de efecto adverso, y la mayoría de los fármacos que ingresan a los ensayos nunca benefician a nadie. Por definición, aún no existen datos en humanos que sugieran que el siRNA divalente beneficiará a alguien con enfermedad priónica. Cualquier persona que se ofrezca como voluntaria para un ensayo, este o cualquier otro ensayo, debe hacerlo con la expectativa de contribuir a la investigación, no de beneficiarse personalmente.
Segundo, actualmente estamos limitados a administrar una sola dosis del fármaco. Esto se debe a que, hasta la fecha, solo tenemos datos de toxicología de dosis única en animales. Estamos trabajando en el lanzamiento de estudios de toxicología crónica, pero aún no conocemos el cronograma ni el resultado. Por lo tanto, en este momento no podemos prometer en absoluto a nadie en el ensayo que alguna vez habrá la posibilidad de recibir otra dosis.
Tercero, no somos un patrocinador comercial, y aunque tenemos la suerte de haber llegado tan lejos, es muy poco probable que podamos encontrar financiación como laboratorio académico para realizar el tipo de ensayos clínicos más amplios que en última instancia conducirían a la aprobación de un fármaco. Por lo tanto, no podemos prometer un futuro potencial en el que este fármaco esté disponible.
Cuarto, nuevamente debido a recursos limitados, aún no hemos explorado la posibilidad de administrar el fármaco a pacientes fuera de los Estados Unidos. Nos encantaría que el siRNA divalente tuviera un futuro global para los pacientes en todas partes. Por hoy, la FDA es la agencia reguladora que mejor conocemos y a la que tenemos el mejor acceso como estadounidenses, así que ahí fue donde fuimos a discutir cómo obtener la autorización para una primera dosis en humanos. Mientras tanto, la financiación del NIH que nos trajo hasta aquí se orientó a satisfacer los requisitos regulatorios de EEUU, y la financiación del NIH que hemos solicitado solo apoyaría un ensayo en EEUU.
Para todos los puntos anteriores, recuerde que no es que no queramos hacer más. Nuestra elección hasta ahora ha sido entre no hacer nada y hacer algo, y en cada coyuntura, hemos elegido hacer algo. Ahora hablemos de lo que creemos que podemos lograr al realizar un ensayo clínico de siRNA divalente. El objetivo principal de un ensayo será obtener datos preliminares sobre si el fármaco es seguro en las dosis probadas. En segundo lugar, el ensayo evaluará si el fármaco pudo alcanzar su objetivo, es decir, reducir la PrP en el cerebro, en esas mismas dosis. Ningún siRNA divalente para ninguna enfermedad ha estado antes en humanos. Y solo 1 fármaco diseñado para reducir la PrP, el ASO ION717 de Ionis, ha sido administrado antes a humanos. Creemos que un pequeño primer ensayo de siRNA divalente en la enfermedad priónica tiene el potencial de enseñarnos sobre cómo funciona el siRNA divalente en el cuerpo humano (su seguridad, potencia, duración de acción), así como sobre la reducción de PrP en pacientes con enfermedad priónica. Al mismo tiempo, un ensayo también nos enseñará sobre la enfermedad priónica y cómo realizar ensayos clínicos en esta enfermedad. Por ejemplo, qué tipo de pacientes podemos reclutar, en qué etapa de la enfermedad, con qué rapidez progresan en diversas medidas y, quizás, cuál es la cinética de la reducción de PrP en esta población. Aprendemos mucho de cada ensayo que se realiza en nuestra enfermedad, y tenemos mucho más que aprender.
El ensayo global PrProfile de ION717 de Ionis, incluso mientras aún está en curso, nos ha enseñado que nuestra comunidad de pacientes es grande y está motivada: la gente se presentó en tal número que el ensayo tuvo que suspenderse durante 4 meses. Hay muchísimas más lecciones que aprender de los ensayos en la enfermedad priónica, que serán increíblemente valiosas para la próxima persona que diseñe un ensayo de (con suerte) un fármaco aún mejor. Aprender algunas de esas lecciones en el contexto de un estudio iniciado por un investigador es especialmente valioso, porque nosotros, como académicos y especialmente como pacientes-científicos, podemos optar por compartir datos en la mayor medida compatible con la privacidad del paciente, con el fin de maximizar el conocimiento generalizable y el beneficio para toda nuestra comunidad de investigación. Sin duda, nosotros, la comunidad de pacientes, podemos y debemos presionar a las empresas para que compartan más datos públicamente, y hasta cierto punto nos escucharán, pero las empresas que cotizan en bolsa también tienen la obligación legal de hacer lo correcto por sus accionistas y, siendo realistas, nunca van a compartir tanto como queremos.
Finalmente, aunque todavía es difícil predecir el desenlace, existe la posibilidad de que el siRNA divalente tenga un papel importante en el tratamiento de la enfermedad priónica, siempre que los resultados del primer ensayo en humanos sean prometedores. Cabe destacar que obtener buenos resultados en modelos murinos es relativamente frecuente, pero lo verdaderamente decisivo son los datos obtenidos en pacientes. Si los resultados iniciales en humanos demuestran ser positivos, esto podría atraer el interés de un patrocinador con mayores recursos que los nuestros, capaz de impulsar su desarrollo hasta convertirlo en un medicamento aprobado. No olvidemos que el objetivo último de toda nuestra investigación es conseguir un fármaco seguro y eficaz tanto para el tratamiento como para la prevención de la enfermedad priónica.
¿Qué ocurre si dos ensayos están reclutando simultáneamente?
En este momento, el ensayo PrProfile de Ionis ha cerrado la inscripción, e Ionis aún no ha anunciado un próximo ensayo de ION717. Pero es posible que haya una superposición entre un futuro ensayo de ION717 y un futuro ensayo de siRNA divalente. ¿Por qué necesitamos más de un fármaco en desarrollo clínico? Porque queremos maximizar la posibilidad de obtener, en última instancia, uno o más fármacos seguros y eficaces. También queremos maximizar los conocimientos de investigación que obtenemos en el camino. La mayoría de los fármacos fallan, e incluso cuando tienen éxito, es raro que el primer fármaco en una enfermedad previamente intratable sea el único fármaco que alguien necesite alguna vez. Por lo general, se necesitan muchos esfuerzos diferentes para finalmente convertir una enfermedad uniformemente fatal en una condición manejable o prevenible. Necesitamos múltiples oportunidades de gol.
¿Está bien tener más de un fármaco en desarrollo clínico? Sí. Solo en Estados Unidos se diagnostican 500 pacientes con enfermedad priónica cada año. Probablemente se diagnostican pocos miles de casos cada año en todo el mundo. El ensayo PrProfile de Ionis solo inscribió a 56 pacientes en todo el mundo. La inscripción espectacularmente rápida de ese ensayo demuestra que los pacientes existen y están muy motivados. Desafortunadamente, tenemos pacientes más que suficientes para inscribir ensayos de múltiples fármacos al mismo tiempo. Y desafortunadamente, la mayoría de los pacientes mueren muy rápidamente, lo que significa que inscribir pacientes este año no «agota» la población de pacientes para el próximo año.
Si los pacientes son elegibles para más de un ensayo, ¿cómo elegirán en cuál inscribirse? Esa es una conversación que deben tener con su médico; no puedo aconsejarle al respecto. Ya he compartido anteriormente algunas de las que creo que son las mayores advertencias sobre nuestro fármaco. Cuando un ensayo finalmente esté reclutando, habrá un formulario de consentimiento que le dará mucho más en qué pensar. Sobre todo, recuerde que esto es investigación. Al momento de escribir esto, no hay datos en humanos que sugieran que algún fármaco sea eficaz contra la enfermedad priónica.
Transparencia
Como pacientes-científicos, queremos maximizar la investigación sobre la enfermedad priónica, y eso significa un compromiso de compartir nuestros conocimientos y datos en la mayor medida posible. Si ha seguido este blog, sabe que hemos insistido mucho en esto durante años, pero dado que ahora estamos en una nueva etapa donde los ensayos en humanos podrían estar cerca, aquí hay un nuevo anticipo de ese compromiso.
NOTA: En esta sección se comparten detalles técnicos sobre el potencial fármaco. Aunque no se reproducen aquí para facilitar la lectura de esta versión adaptada, toda la información está públicamente disponible en el artículo original en cureffi.org.
Conclusiones
Desarrollamos un nuevo fármaco candidato para la enfermedad priónica en nuestro laboratorio, obtuvimos permiso de la FDA para administrar el fármaco a pacientes humanos y ahora estamos buscando la financiación para lanzar un ensayo clínico. El ensayo aún no se ha lanzado. No me escriban preguntando cómo pueden obtener el fármaco: todavía no hay manera. Estamos trabajando duro en esto. Si puede ayudarnos a encontrar la financiación, háganoslo saber. Cuando lleguemos a un ensayo, seguirá siendo investigación, no tratamiento, y nadie debería participar con la expectativa de que les ayudará. Si desea mantenerse informado sobre los nuevos desarrollos, siga este blog y, mientras lo hace, únase a nuestra lista de correo y considere unirse a PrionRegistry si desea ser voluntario para la investigación. Manténganse atentos.