Autor: Hasier Eraña
Un equipo de investigadores estadounidenses ha publicado en la prestigiosa revista Nature una técnica revolucionaria de edición genética (técnica que permite introducir cambios en el material genético de las células) que podría cambiar la forma en que abordamos el tratamiento de diversas enfermedades genéticas o de origen familiar de baja prevalencia, también conocidas como enfermedades raras, de las que actualmente se han descrito hasta 7000 enfermedades distintas. El nuevo método, denominado PERT (por sus siglas en inglés, “edición dirigida por prime editing para leer a través de codones de terminación prematuros”), ha demostrado ser capaz de restaurar la función de proteínas en enfermedades tan diversas como la fibrosis quística, la enfermedad de Tay-Sachs o el síndrome de Hurler utilizando exactamente el mismo tratamiento. Esta capacidad de tratar múltiples enfermedades familiares con causas diferentes con una única composición terapéutica representa un cambio radical respecto a las estrategias actuales de medicina de precisión.
¿En qué consiste esta nueva técnica?
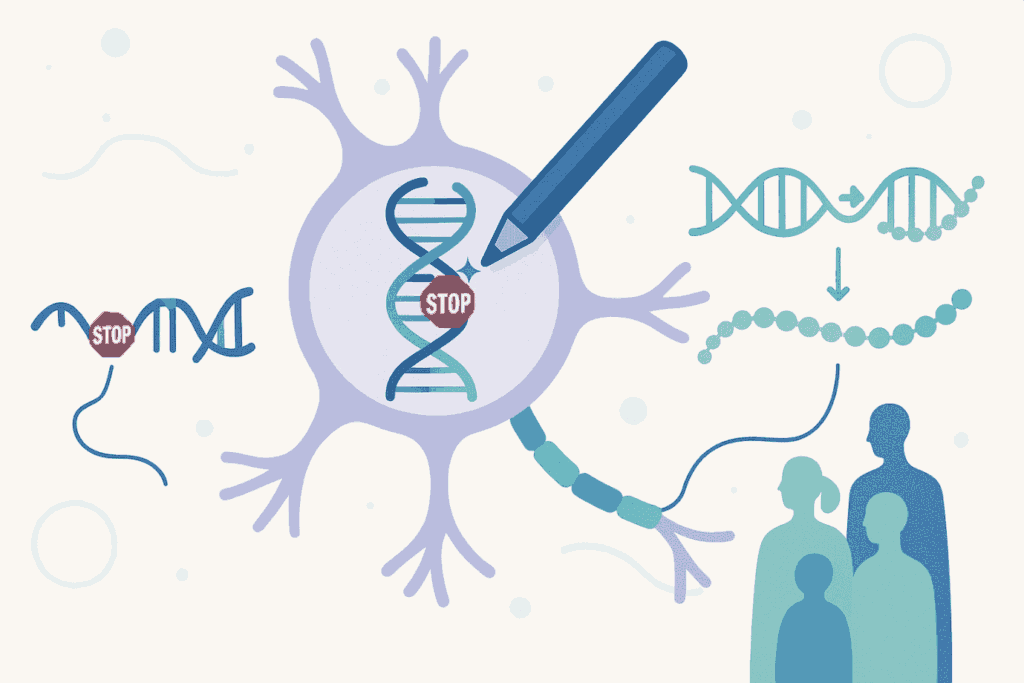
Aproximadamente una cuarta parte de las enfermedades genéticas o familiares están causadas por “mutaciones sin sentido” o “nonsense mutations” en un gen particular. Los genes, constituyen las instrucciones para la fabricación de proteínas en las células. Las mutaciones sin sentido introducen señales de parada prematura en las instrucciones genéticas, haciendo que las células dejen de fabricar una proteína antes de que esté completa. El resultado es una proteína truncada e inactiva que no puede realizar su función.
La estrategia PERT utiliza una tecnología de edición genética avanzada llamada “prime editing” para convertir de forma permanente un gen de ARN de transferencia (tRNA) redundante en el genoma en un tRNA supresor optimizado. Los tRNAs son moléculas esenciales que ayudan a traducir la información genética en proteínas, es decir, son transportadores que enlazan las instrucciones genéticas con los componentes necesarios para fabricar las proteínas. Los tRNAs supresores son versiones especiales de estos transportadores, capaces de “ignorar” esas señales de parada prematuras, permitiendo que la célula complete la fabricación de la proteína funcional, a pesar de la mutación o error en sus instrucciones genéticas.
Lo más innovador de PERT es que, con una única modificación genética administrada una sola vez (dirigida a modificar el tRNA), se puede tratar a pacientes con mutaciones sin sentido en genes completamente diferentes. Es decir, el mismo tratamiento podría beneficiar teóricamente a personas con fibrosis quística, distrofia muscular de Duchenne, fenilcetonuria o enfermedad de Stargardt, entre muchas otras, siempre que su enfermedad esté causada por este tipo concreto de mutación.
¿Qué resultados ha mostrado el estudio?
Los investigadores han llevado a cabo una exhaustiva serie de experimentos que demuestran la eficacia de PERT en modelos celulares y modelos animales:
En modelos celulares humanos de enfermedades lisosomales (enfermedad de Batten, Tay-Sachs y Niemann-Pick tipo C), el tratamiento logró restaurar entre un 20% y un 70% de la actividad normal de las proteínas implicadas. Estos niveles están muy por encima del umbral terapéutico necesario para mejorar significativamente estas enfermedades.
En ratones con el síndrome de Hurler, una grave enfermedad de almacenamiento lisosomal, PERT restauró aproximadamente un 6% de la actividad de la proteína deficiente, lo que resultó en un rescate casi completo de todas las manifestaciones de la enfermedad.
Los investigadores también probaron PERT frente a más de 14.700 mutaciones sin sentido patogénicas registradas en bases de datos clínicas, observando que la técnica fue capaz de leer a través de la gran mayoría de ellas.
Crucialmente, los exhaustivos análisis de seguridad no detectaron lectura a través de codones de parada naturales (que sí deben funcionar correctamente, ya que indican cuándo debe terminar de fabricarse una proteína), ni alteraciones en el transcriptoma o proteoma celular, ni edición fuera de lugar. Los ratones tratados tampoco mostraron signos de toxicidad debida al tratamiento durante el seguimiento de 15 semanas.
¿Qué implicaciones podría tener para las enfermedades priónicas y otras enfermedades neurodegenerativas?
Si bien es importante señalar que muy pocas enfermedades priónicas u otras enfermedades neurodegenerativas están causadas por mutaciones sin sentido (ya que la mayoría se deben a mutaciones de cambio de sentido u otras alteraciones), este trabajo representa un avance conceptual importante para el campo de la terapia génica y las enfermedades raras en general.
La tecnología de “prime editing” empleada por PERT es extremadamente versátil y precisa, y podría adaptarse en el futuro para abordar otros tipos de mutaciones genéticas. De hecho, varios grupos de investigación, incluido nuestro laboratorio, están explorando activamente el uso de prime editing y otras tecnologías similares de edición génica para desarrollar terapias dirigidas específicamente a las mutaciones causantes de enfermedades priónicas familiares.
Además, el concepto de desarrollar terapias “independientes de la enfermedad” que puedan tratar a múltiples pacientes con distintas patologías utilizando una única composición es especialmente relevante para las enfermedades raras como las priónicas, donde el número limitado de pacientes dificulta el desarrollo de terapias específicas para cada mutación.
En resumen
Este estudio representa un hito importante en el campo de la medicina de precisión al demostrar que es posible desarrollar terapias genéticas que no necesitan ser diseñadas individualmente para cada mutación, sino que pueden beneficiar a pacientes con múltiples enfermedades diferentes. Aunque se trata todavía de investigación preclínica y serán necesarios años de estudios adicionales antes de que pueda aplicarse en humanos, PERT abre la puerta a un nuevo paradigma terapéutico en el que un único medicamento podría transformar el tratamiento de cientos de enfermedades genéticas, beneficiando potencialmente a cientos de miles de pacientes en todo el mundo.
Enlace al artículo original.