Auteur : Hasier Eraña
Du laboratoire de Sonia Vallabh et Eric Minikel, au Broad Institute de Boston, vient une nouvelle qu'Eric lui-même rapporte sur son blog (cureffi.org). Le 14 avril, ces chercheurs ont franchi une étape rare dans la recherche universitaire (celle qui se fait en dehors de l'industrie pharmaceutique), à savoir l'autorisation d'un nouveau médicament expérimental (IND) par la Food and Drug Administration américaine (FDA, l'agence responsable de l'autorisation des essais de nouveaux médicaments sur l'homme). Il s'agit d'une étape importante sur le chemin long et coûteux qui mène de la découverte au développement et à la commercialisation d'une nouvelle thérapie, en l'occurrence une nouvelle approche visant à réduire la PrPC dans le cerveau à l'aide d'un siRNA divalent. Bien que les chercheurs eux-mêmes reconnaissent qu'il reste un long chemin à parcourir pour prouver l'efficacité et la sécurité de ce nouveau traitement chez les patients atteints de maladies à prions, ils ont voulu célébrer la bonne nouvelle dans un article de blog complet, détaillant l'état actuel du développement de cette thérapie potentielle et décrivant les prochaines étapes du processus. Je résume ci-dessous le contenu de l'article publié par Eric Minikel le 14 avril, dont la version originale est disponible à l'adresse suivante cureffi.org.
Le parcours d'un candidat-médicament
En 2019, le Dr Anastasia Khvorova, de l'école de médecine de l'université du Massachusetts, s'est rendue au Broad Institute pour parler de son parcours professionnel dans la conception de meilleurs médicaments à base d'ARN interférent court, ou siRNA. Elle s'est concentrée sur une étape très récente : l'invention par son laboratoire de ce qu'ils appellent le siRNA divalent.
Jusqu'alors, les oligonucléotides antisens (ASO) étaient la seule option pour réduire une protéine cible dans le cerveau (ils font maintenant l'objet d'un essai de phase I grâce à une collaboration avec Ionis Pharmaceuticals). Mais nous avions besoin d'une autre chance d'atteindre l'objectif. La publication d'Anastasia sur le siRNA divalent montrait une réduction très profonde et durable d'un gène cible (HTT), et nous avons pensé que si nous pouvions faire la même chose avec notre gène, le PRNP, ce serait vraiment prometteur.
Son équipe a rapidement sélectionné des séquences de siRNA contre des gènes humains et de souris, et a déposé un brevet pour plusieurs d'entre elles [WO2021173984]. En combinant les données préliminaires de son laboratoire sur les séquences d'ARNsi et les données préliminaires de notre laboratoire sur les nouveaux modèles murins de la PrP humaine et sur un test permettant de quantifier la déplétion de la PrP dans le cerveau, nous avons demandé une subvention aux National Institutes of Health (NIH) pour soutenir le développement d'un médicament. À notre grande surprise, nous l'avons obtenue.
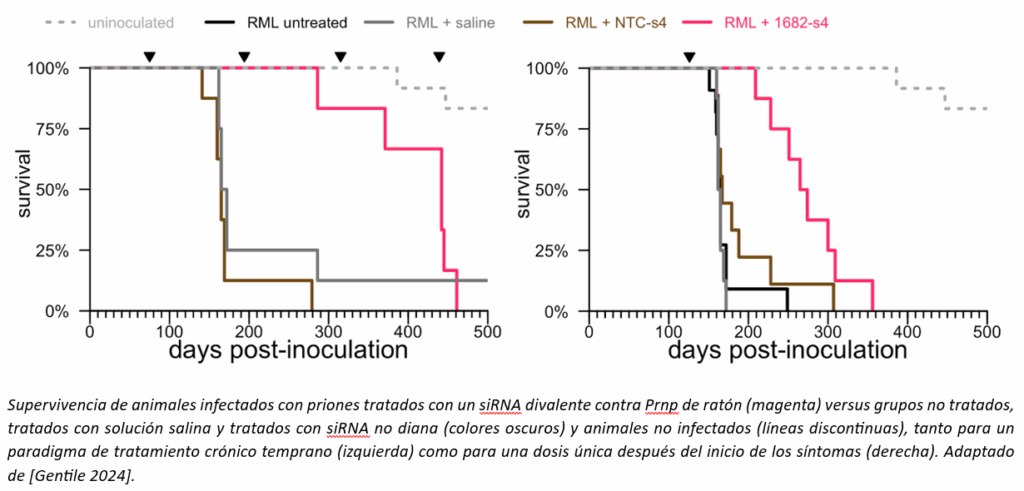
Nous avons testé les molécules de siRNA envoyées par l'université du Massachusetts, d'abord sur des cellules puis sur des souris, afin de trouver la meilleure molécule. Le meilleur siRNA que nous avons trouvé contre la Prnp de la souris n'était pas exceptionnellement puissant ; il n'a réduit la PrP dans l'ensemble du cerveau de la souris que d'environ 50%. Mais cela a suffi pour poser la question suivante : la réduction de la PrP à l'aide d'un siRNA divalent améliore-t-elle la survie, comme nous l'avions constaté avec les OLS ? Il s'agit d'expériences à long terme, dans le cadre desquelles un groupe d'animaux doit être infecté par des prions et, des mois plus tard, recevoir le médicament à un moment très précis. L'expérience a fonctionné : le traitement par l'ARNsi divalent, même une seule dose juste après l'apparition des symptômes, a fait une grande différence dans la durée de vie des animaux infectés par les prions.
Entre-temps, à l'automne 2022, nous avions choisi un composé siRNA divalent unique contre la PRNP humaine qui, d'après toutes les expériences que nous avions pu mener, était le meilleur de tous. Nous avons constaté une réduction de la PrP d'une profondeur jamais vue auparavant, avec une durabilité de plusieurs mois, et il semblait être bien toléré. Nous voulions désespérément qu'il s'agisse d'un autre cheval dans la course au traitement de la maladie à prions. Les choix étaient clairs : soit nous développions nous-mêmes ce médicament, soit nous acceptions la dure réalité que tout ce travail ne serait qu'une "simple étude sur des souris", un énième article scientifique qui dit "cela semble prometteur" mais qui n'arrivera jamais dans la vie d'un être humain.
À peu près au même moment, nous avons appris l'existence d'un nouveau mécanisme de financement des NIH, un programme axé précisément sur notre situation : vous disposez de données prometteuses chez la souris, mais il faudra des années de travail et des millions de dollars pour obtenir une première dose chez l'homme. Ce programme semblait être notre seule chance, mais il n'y avait aucun doute : même avec le soutien financier des NIH pour rendre la tâche réalisable, cela allait représenter un effort très, très important pour nous.
Une parenthèse scientifique : comment fonctionne le siRNA divalent ?
SiRNA est l'abréviation de short interfering RNA (ARN interférent court). Ce type de médicament est composé des mêmes éléments que l'ADN et l'ARN (A, C, G et T ou U) et agit par le mécanisme de l'interférence ARN (ARNi). L'ARNi a été découvert en 1998 et a valu à ses découvreurs le prix Nobel de physiologie ou de médecine en 2006. Le médicament siRNA est conçu pour se lier spécifiquement à une molécule d'ARN cible dans la cellule, en l'occurrence celle qui donne naissance à la PrP. Il en résulte une diminution de l'ARN cible et donc de la protéine qu'il code. Un siRNA est donc un moyen de réduire la quantité d'une protéine spécifique responsable d'une maladie. Le premier médicament à base de siARN, Patissiran pour l'amylose à transthyrétine, a reçu l'approbation de la FDA en 2018.
Un siRNA est similaire à un ASO en ce sens qu'il s'agit d'un médicament conçu pour se lier à un ARN cible et le dégrader. Comme un ASO, chez l'homme, ce siRNA serait administré dans le liquide céphalorachidien (LCR) par une injection intrathécale, autrement dit une ponction lombaire (PL). Un siRNA diffère d'un ASO en ce sens qu'il s'agit d'un oligonucléotide double brin (et non simple brin), qu'il utilise des schémas de modification chimique différents pour le stabiliser et qu'il agit par le biais d'une voie cellulaire différente (RISC et non ARNase H1).
L'année dernière, six médicaments à base de siARN avaient été approuvés par la FDA et des dizaines d'autres étaient en cours d'essais cliniques. Plusieurs sociétés pharmaceutiques développent des siRNA avec différentes modifications chimiques, différents conjugués pour favoriser l'absorption dans différents tissus du corps, et différentes séquences pour différentes cibles dans différentes indications de maladies.
Un siRNA divalent est un type spécial développé par le laboratoire d'Anastasia Khvorova. Il se compose de deux copies du même siRNA à double brin, reliées entre elles. Des études empiriques menées sur des souris par le laboratoire d'Anastasia Khvorova lors de la mise au point de cette technologie ont montré que les siARN simples (monovalents), avec leur chimie particulière, n'étaient tout simplement pas absorbés et retenus dans le cerveau. Lorsqu'ils en ont réuni deux pour former une molécule divalente, une grande quantité de médicament a soudain été retenue dans le cerveau, a été absorbée par les cellules et s'est révélée pharmacologiquement active.
L'ascension de la montagne IND (Investigational New Drug)
Aux États-Unis, et en fait dans tous les pays du monde, les étapes nécessaires pour passer de "ça a l'air bon sur une souris" à l'autorisation d'administrer un médicament à une personne ne sont pas seulement longues et coûteuses, ce sont aussi des études hautement spécialisées. Il existe toute une industrie que, si vous ne travaillez pas dans le développement de médicaments, vous n'avez jamais vue ou à laquelle vous n'avez jamais pensé, et qui est gérée par des entreprises dont vous ne connaissez pas le nom. La synthèse chimique, la purification, les tests de qualité et le conditionnement des médicaments (chimie, fabrication et contrôles, CMC, dans le jargon de l'industrie) doivent être réalisés selon les bonnes pratiques de fabrication (BPF), ce qui est dix fois plus coûteux que la fabrication d'un médicament destiné à être utilisé chez la souris. Tout d'abord, toutes les étapes des BPF sont suivies pour fabriquer une substance médicamenteuse (SD), une poudre blanche, puis un processus supplémentaire complet est suivi pour formuler cette poudre en un produit médicamenteux (MP), qui peut être une pilule ou, dans notre cas, un flacon de liquide injectable stérile. Les études toxicologiques doivent être menées selon les bonnes pratiques de laboratoire (BPL), avec des dizaines ou des centaines d'animaux, chacun produisant des centaines, voire des milliers de mesures et de lectures pour déterminer si le médicament est toxique. Le sang et/ou les tissus des animaux sont analysés à l'aide d'un test pharmacocinétique validé par les BPL afin de confirmer la quantité de médicament réellement présente et de s'assurer que le dosage a été effectué correctement. Des études d'interaction médicamenteuse (DDI) sont menées pour déterminer si les patients qui prennent l'un des centaines de médicaments approuvés devraient se voir interdire l'utilisation de leur médicament. Des études de génotoxicité permettent de déterminer si le médicament peut entraîner une mutation de l'ADN et provoquer un cancer ou des malformations congénitales.
L'intérêt de tout ceci est d'en venir à un autre acronyme : l'IND, ou Investigational New Drug (nouveau médicament expérimental). Une demande d'IND est un ensemble de données provenant de toutes les études antérieures qu'un développeur ou un promoteur de médicaments soumet à la FDA pour obtenir l'autorisation d'administrer un nouveau médicament à un être humain pour la première fois.
Comme nous avons soumis notre proposition au NIH en octobre 2022, nous savions qu'il faudrait attendre près d'un an avant que le financement ne soit accordé, et probablement un peu plus tard avant que les contrats ne soient établis et que le travail ne puisse commencer sérieusement. Désireux d'aller de l'avant le plus rapidement possible, nous avons commencé à fabriquer le médicament immédiatement grâce au financement de Prion Alliance. En juin 2023, Sonia et moi avons eu notre première réunion avec la FDA, au cours de laquelle nous avons pu poser des questions sur ce qui serait nécessaire pour notre IND compte tenu des particularités de notre médicament et de notre maladie. Avec l'avis de la FDA en main, nous avons travaillé avec des donateurs extrêmement intelligents, dévoués et passionnés afin de collecter des fonds pour certaines des autres étapes les plus contraignantes, en particulier la première série d'études toxicologiques et le développement d'un essai pharmacocinétique, la méthode de quantification du médicament dans le sang ou les tissus.
Nos ressources financières étaient encore assez limitées et nous avons estimé que la rapidité était essentielle, c'est pourquoi nous avons fait des compromis difficiles. Nous avons opté pour une étude toxicologique à dose unique sur les animaux, ce qui, la FDA nous l'a clairement fait comprendre, ne nous permettrait de mener qu'un essai à dose unique sur les patients. Nous avons pris cette décision pour plusieurs raisons. Nous ne pouvions nous permettre qu'une étude à dose unique, et une étude à dose unique serait plus rapide. Nos données chez la souris ont montré qu'une dose unique pouvait avoir une activité de 4 à 6 mois, et qu'une dose unique administrée à un stade symptomatique de la maladie prolongeait de 3,5 mois la survie des souris infectées par le prion. Par conséquent, étant donné que de nombreux patients atteints de la maladie à prion ne vivent que quelques mois après le diagnostic, nous avons estimé que même une dose unique pouvait être cliniquement pertinente. Enfin, une étude toxicologique à dose unique présentait moins de risques. En règle générale, les études toxicologiques sont déjà menées avec plusieurs fois l'équivalent de la dose que l'on s'attend à utiliser chez l'homme. Si l'on ajoute à cela une administration mensuelle pendant 9 mois (comme c'est généralement le cas dans les études de toxicité chronique), les animaux reçoivent une très grande quantité de médicament, ce qui augmente considérablement la probabilité d'effets toxiques qui empêcheraient de passer aux essais cliniques.
Par un incroyable coup de chance, notre proposition de subvention aux NIH a finalement été financée. Cela nous a permis d'aller de l'avant et de mener toutes les études restantes nécessaires à l'obtention d'une IND. Ce processus a nécessité deux ans et demi de travail intense, mené conjointement et en grande partie sous la direction d'Alissa Coffey, chef de projet.
Au début de l'année 2025, toutes les études d'habilitation des IND étaient terminées et il ne restait plus qu'à rédiger l'IND proprement dite. Bien que nous ayons fait appel à des consultants, trouver des modèles et des exemples à partir desquels travailler a constitué un défi majeur. L'industrie conduit ses IND selon une norme industrielle, qui va même au-delà de ce qui est exigé pour une IND initiée par un chercheur. Les universitaires qui ont soumis des IND, et il n'y a pas beaucoup d'exemples pour commencer, ne l'ont généralement pas fait pour de nouvelles modalités médicamenteuses, telles que l'ARNsi divalent. Et les IND disponibles, qu'elles soient industrielles ou universitaires, sont très difficiles à trouver. Mais, très généreusement, certaines personnes ont partagé leurs IND avec moi, ce qui m'a permis d'avoir une sorte de point de départ pour travailler. Le 5 février 2025, Alissa et moi nous sommes assis dans mon bureau et avons cliqué sur "envoyer".
Ce que signifie notre IND ouverte
Lorsque la FDA approuve une IND, elle n'approuve pas seulement un médicament, elle approuve un lot spécifique de médicaments pour une population spécifique de patients, traités selon un protocole d'essai clinique spécifique. Chaque détail doit être spécifié. Dans notre IND, nous avons soumis deux protocoles d'essai : l'un pour traiter les patients symptomatiques déjà diagnostiqués avec une maladie à prion, et l'autre pour traiter les patients pré-symptomatiques présentant un risque de maladie à prion. Le 14 mars, la FDA a répondu que "nous pouvons continuer" pour les patients symptomatiques, alors que nous sommes en "attente clinique partielle" pour les patients présymptomatiques. Cela signifie qu'elle souhaite obtenir davantage de données avant de nous autoriser à administrer des doses aux patients présymptomatiques, et elle nous a envoyé une lettre détaillée décrivant tout ce que nous devrons faire pour débloquer l'administration de doses aux patients présymptomatiques.
Mais en attendant, nous avons une IND ouverte : la FDA nous autorise à démarrer un essai clinique sur des patients symptomatiques atteints de la maladie à prion. J'aimerais que cela signifie que nous pourrions commencer à administrer des doses aux patients dès demain, mais la réalité est plus compliquée. Tout d'abord, nous ne disposons pas encore des fonds nécessaires à la réalisation d'un essai clinique. Le NIH dispose également d'un programme à cet effet et, après avoir passé avec succès l'étape 1 de ce mécanisme de financement, et quelques jours après avoir reçu une réponse de la FDA, nous avons soumis notre proposition pour l'étape 2. Nous ne savons toujours pas si et quand ce projet sera financé. Au plus tôt, cela pourrait se produire dans quelques mois. Mais cela pourrait aussi prendre plusieurs mois, voire jamais.
Supposons que quelqu'un nous fasse aujourd'hui un chèque correspondant au coût total d'un essai. Même dans ce cas, il faudrait du temps pour le mettre en place et le faire fonctionner. Un essai clinique est une entreprise extrêmement complexe. De plus, si le NIH finance l'essai, il déterminera divers détails sur la manière dont l'essai sera mené, et une fois l'approbation éthique obtenue, il y aura encore une longue liste d'exigences à respecter pour la réalisation de l'essai. les tâches à accomplir.
Je suis plus impatient que quiconque de savoir quand nous commencerons à administrer des doses aux patients, mais il y a de grandes inconnues pour le moment, notamment le fait que nous ne pouvons pas encore exclure que la réponse soit "jamais" si nous ne parvenons pas à trouver le financement. Lorsque j'ai parlé à des personnes ayant l'expérience des premiers essais sur l'homme, la plupart m'ont dit que, si le financement arrive rapidement, il serait possible d'administrer le médicament à un premier patient d'ici à la fin de l'année 2025.
À quoi s'attendre lorsque nous serons prêts à administrer le médicament aux patients ?
Si le financement le permet, nous pourrons, au cours des prochains mois, annoncer qu'un essai clinique est ouvert et commencer à accepter des patients. Voici quelques éléments à garder à l'esprit lorsque cela se produira :
Tout d'abord, un essai clinique est une recherche, pas un traitement. Nous menons un essai clinique précisément parce que nous ne savons pas encore si le médicament est sûr ou efficace. Tous les médicaments provoquent un certain type d'effet indésirable et la plupart des médicaments qui sont soumis à des essais ne profitent jamais à personne. Par définition, il n'existe pas encore de données humaines suggérant que le siRNA divalent sera bénéfique à toute personne atteinte d'une maladie à prions. Quiconque se porte volontaire pour un essai, celui-ci ou tout autre essai, doit le faire dans l'espoir de contribuer à la recherche et non d'en tirer un bénéfice personnel.
Deuxièmement, nous sommes actuellement limités à l'administration d'une seule dose du médicament. En effet, à ce jour, nous ne disposons que de données toxicologiques à dose unique chez l'animal. Nous travaillons au lancement d'études de toxicologie chronique, mais nous n'en connaissons pas encore le calendrier ni les résultats. Par conséquent, à ce stade, nous ne pouvons absolument pas promettre aux participants à l'essai qu'ils auront un jour la possibilité de recevoir une autre dose.
Troisièmement, nous ne sommes pas un sponsor commercial et, bien que nous ayons la chance d'être arrivés jusqu'ici, il est très improbable que nous puissions trouver un financement en tant que laboratoire universitaire pour mener le type d'essais cliniques plus importants qui conduiraient finalement à l'approbation d'un médicament. Par conséquent, nous ne pouvons pas promettre un futur potentiel dans lequel ce médicament sera disponible.
Quatrièmement, toujours en raison de ressources limitées, nous n'avons pas encore exploré la possibilité d'administrer le médicament à des patients en dehors des États-Unis. Nous aimerions que le siRNA divalent ait un avenir mondial pour les patients du monde entier. Pour l'instant, la FDA est l'agence réglementaire que nous connaissons le mieux et à laquelle nous avons le plus accès en tant qu'Américains, et c'est donc à elle que nous nous sommes adressés pour discuter de la manière d'obtenir l'approbation d'une première dose chez l'homme. En attendant, le financement des NIH qui nous a permis d'arriver ici était destiné à répondre aux exigences réglementaires américaines, et le financement des NIH que nous avons sollicité ne permettrait de financer qu'un seul essai aux États-Unis.
Pour tous les points susmentionnés, n'oubliez pas que ce n'est pas parce que nous ne voulons pas en faire plus. Jusqu'à présent, nous avons eu le choix entre ne rien faire et faire quelque chose, et à chaque fois, nous avons choisi de faire quelque chose. Parlons maintenant de ce que nous pensons pouvoir réaliser en menant un essai clinique avec un siRNA divalent. L'objectif premier d'un essai sera d'obtenir des données préliminaires sur l'innocuité du médicament aux doses testées. Deuxièmement, l'essai évaluera si le médicament est capable d'atteindre son objectif, c'est-à-dire de réduire la PrP dans le cerveau, à ces mêmes doses. Aucun siRNA divalent pour quelque maladie que ce soit n'a jamais été administré à l'homme auparavant. Et seul un médicament conçu pour réduire la PrP, l'ASO ION717 de Ionis, a déjà été administré à des humains. Nous pensons qu'un premier essai à petite échelle du siRNA divalent dans la maladie de prion peut nous apprendre comment le siRNA divalent fonctionne dans le corps humain (sa sécurité, sa puissance, sa durée d'action), ainsi que sur la réduction de la PrP chez les patients atteints de la maladie de prion. Dans le même temps, un essai nous permettra également d'en savoir plus sur la maladie à prions et sur la manière de mener des essais cliniques dans cette maladie. Par exemple, quel type de patients nous pouvons recruter, à quel stade de la maladie, à quelle vitesse ils progressent selon diverses mesures, et peut-être quelle est la cinétique de la réduction de la PrP dans cette population. Nous apprenons beaucoup de chaque essai réalisé dans notre maladie, et nous avons encore beaucoup à apprendre.
L'essai mondial PrProfile de Ionis sur le ION717, même s'il est encore en cours, nous a appris que notre communauté de patients est nombreuse et motivée : les gens se sont présentés en si grand nombre que l'essai a dû être suspendu pendant quatre mois. Il y a beaucoup, beaucoup d'autres leçons à tirer des essais sur les maladies à prions, qui seront incroyablement précieuses pour la prochaine personne qui concevra un essai sur un médicament (espérons-le) encore meilleur. Apprendre certaines de ces leçons dans le contexte d'une étude menée à l'initiative d'un chercheur est particulièrement précieux, car nous, en tant qu'universitaires et surtout en tant que patients-chercheurs, pouvons choisir de partager les données dans toute la mesure compatible avec la vie privée des patients, afin de maximiser les connaissances généralisables et les avantages pour l'ensemble de notre communauté de recherche. Certes, nous, la communauté des patients, pouvons et devons pousser les entreprises à partager davantage de données publiquement, et dans une certaine mesure, elles nous écouteront, mais les entreprises cotées en bourse ont également l'obligation légale de faire ce qu'il faut pour leurs actionnaires et, de manière réaliste, elles ne partageront jamais autant de données que nous le voudrions.
Enfin, bien qu'il soit encore difficile de prédire les résultats, il est possible que le siRNA divalent joue un rôle important dans le traitement de la maladie à prions, à condition que les résultats du premier essai sur l'homme soient prometteurs. Il convient de noter que les bons résultats dans les modèles murins sont relativement fréquents, mais que ce sont les données sur les patients qui sont réellement décisives. Si les premiers résultats chez l'homme s'avèrent positifs, ils pourraient susciter l'intérêt d'un sponsor disposant de ressources plus importantes que les nôtres, qui pourrait être en mesure de pousser le développement jusqu'à l'obtention d'un médicament homologué. N'oublions pas que l'objectif ultime de toutes nos recherches est de mettre au point un médicament sûr et efficace pour le traitement et la prévention de la maladie à prions.
Que se passe-t-il si deux essais sont recrutés simultanément ?
À l'heure actuelle, l'essai PrProfile de Ionis a clôturé le recrutement et Ionis n'a pas encore annoncé d'essai ION717 à venir. Mais il pourrait y avoir un chevauchement entre un futur essai ION717 et un futur essai siRNA divalent. Pourquoi avons-nous besoin de plus d'un médicament en développement clinique ? Parce que nous voulons maximiser les chances d'obtenir un ou plusieurs médicaments sûrs et efficaces. Nous voulons également maximiser les connaissances que nous acquérons en cours de route. La plupart des médicaments échouent, et même lorsqu'ils réussissent, il est rare que le premier médicament contre une maladie auparavant incurable soit le seul dont on aura jamais besoin. Il faut généralement de nombreux efforts différents pour transformer une maladie uniformément mortelle en une condition gérable ou évitable. Nous avons besoin de multiples opportunités d'objectifs.
Est-il acceptable d'avoir plus d'un médicament en développement clinique ? Oui. 500 patients sont diagnostiqués avec une maladie à prions chaque année rien qu'aux États-Unis. Il y a probablement quelques milliers de cas diagnostiqués chaque année dans le monde. L'essai PrProfile de Ionis n'a recruté que 56 patients dans le monde. Le recrutement extrêmement rapide de cet essai montre que les patients existent et qu'ils sont très motivés. Malheureusement, nous avons plus de patients qu'il n'en faut pour recruter plusieurs essais de médicaments en même temps. Et malheureusement, la plupart des patients meurent très rapidement, ce qui signifie que le recrutement de patients cette année n'épuise pas la population de patients pour l'année prochaine.
Si les patients sont éligibles à plus d'un essai, comment choisiront-ils celui dans lequel ils s'inscriront ? C'est une conversation qu'ils doivent avoir avec leur médecin ; je ne peux pas les conseiller à ce sujet. J'ai déjà fait part de ce que j'estime être les principales mises en garde concernant notre médicament. Lorsqu'un essai sera enfin recruté, il y aura un formulaire de consentement qui vous donnera beaucoup plus d'éléments à prendre en compte. Avant tout, n'oubliez pas qu'il s'agit d'une recherche. Au moment où j'écris ces lignes, il n'existe aucune donnée humaine suggérant qu'un médicament est efficace contre la maladie à prions.
Transparence
En tant que patients-scientifiques, nous voulons maximiser la recherche sur les maladies à prions, ce qui implique un engagement à partager nos connaissances et nos données autant que possible. Si vous suivez ce blog, vous savez que nous insistons sur ce point depuis des années, mais étant donné que nous sommes maintenant à un nouveau stade où les essais sur l'homme pourraient être proches, voici un nouvel aperçu de cet engagement.
NOTE : Cette section présente des détails techniques sur le médicament potentiel. Bien qu'elles ne soient pas reproduites ici pour faciliter la lecture de cette version adaptée, toutes les informations sont accessibles au public dans l'article original à l'adresse suivante cureffi.org.
Conclusions
Nous avons développé un nouveau médicament candidat contre les maladies à prions dans notre laboratoire, obtenu l'autorisation de la FDA d'administrer le médicament à des patients humains et recherchons à présent des fonds pour lancer un essai clinique. L'essai n'a pas encore été lancé. Ne m'écrivez pas pour me demander comment vous pouvez obtenir le médicament : il n'y a pas encore de moyen. Nous y travaillons d'arrache-pied. Si vous pouvez nous aider à trouver le financement, faites-le nous savoir. Lorsque nous en arriverons à un essai, il s'agira toujours de recherche, pas de traitement, et personne ne devrait participer en pensant que cela l'aidera. Si vous souhaitez vous tenir au courant des derniers développements, suivez ce blog et, tant que vous y êtes, inscrivez-vous à notre liste de diffusion et envisagez de vous inscrire au PrionRegistry si vous souhaitez vous porter volontaire pour la recherche. Restez à l'écoute.



