Auteur : Hasier Eraña
Une équipe de chercheurs américains a publié dans la prestigieuse revue Nature une technique révolutionnaire d'édition génétique (technique permettant d'introduire des modifications dans le matériel génétique des cellules) qui pourrait changer la façon dont nous abordons le traitement de diverses maladies génétiques ou d'origine familiale à faible prévalence, également connues sous le nom de maladies rares, dont 7 000 différentes ont été décrites à ce jour. La nouvelle méthode, appelée PERT (pour « prime editing-directed editing to read through premature termination codons », ou édition guidée par prime editing pour lire à travers les codons de terminaison prématurés), s'est avérée capable de restaurer la fonction des protéines dans des maladies aussi diverses que la mucoviscidose, la maladie de Tay-Sachs ou le syndrome de Hurler en utilisant exactement le même traitement. Cette capacité à traiter plusieurs maladies héréditaires ayant des causes différentes avec une seule composition thérapeutique représente un changement radical par rapport aux stratégies actuelles de médecine de précision.
En quoi consiste cette nouvelle technique ?
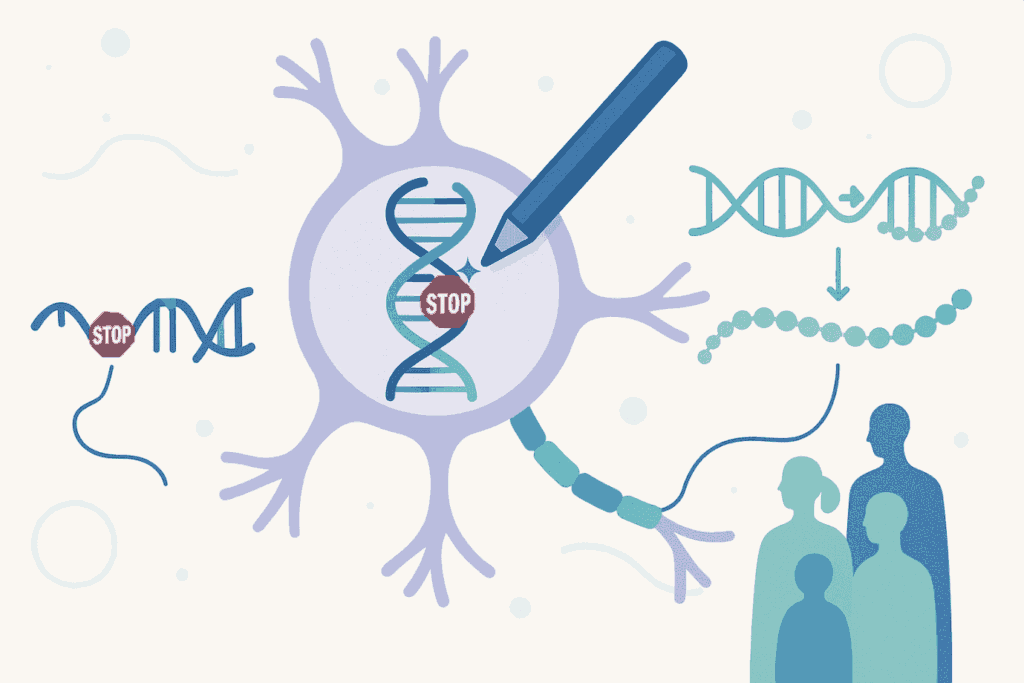
Environ un quart des maladies génétiques ou familiales sont causées par des « mutations non-sens » ou « nonsense mutations » dans un gène particulier. Les gènes constituent les instructions pour la fabrication des protéines dans les cellules. Les mutations non-sens introduisent des signaux d'arrêt prématuré dans les instructions génétiques, ce qui empêche les cellules de fabriquer une protéine avant qu'elle ne soit complète. Il en résulte une protéine tronquée et inactive qui ne peut pas remplir sa fonction.
La stratégie PERT utilise une technologie avancée d'édition génétique appelée « prime editing » pour convertir de manière permanente un gène d'ARN de transfert (ARNt) redondant dans le génome en un ARNt suppresseur optimisé. Les ARNt sont des molécules essentielles qui aident à traduire l'information génétique en protéines, c'est-à-dire qu'ils sont des transporteurs qui relient les instructions génétiques aux composants nécessaires à la fabrication des protéines. Les ARNt suppresseurs sont des versions spéciales de ces transporteurs, capables d'« ignorer » ces signaux d'arrêt prématurés, permettant ainsi à la cellule de terminer la fabrication de la protéine fonctionnelle, malgré la mutation ou l'erreur dans ses instructions génétiques.
La principale innovation du PERT réside dans le fait qu'une seule modification génétique administrée une seule fois (visant à modifier l'ARNt) permet de traiter des patients présentant des mutations non sens dans des gènes complètement différents. En d'autres termes, le même traitement pourrait théoriquement bénéficier aux personnes atteintes de mucoviscidose, de dystrophie musculaire de Duchenne, de phénylcétonurie ou de la maladie de Stargardt, entre autres, à condition que leur maladie soit causée par ce type particulier de mutation.
Quels résultats l’étude a-t-elle montré ?
Les chercheurs ont mené une série exhaustive d'expériences démontrant l'efficacité du PERT sur des modèles cellulaires et animaux :
Dans les modèles cellulaires humains de maladies lysosomales (maladies de Batten, Tay-Sachs et Niemann-Pick de type C), le traitement a permis de restaurer entre 20 % et 70 % de l'activité normale des protéines impliquées. Ces niveaux sont bien supérieurs au seuil thérapeutique nécessaire pour améliorer significativement ces maladies.
Chez les souris atteintes du syndrome de Hurler, une grave maladie de surcharge lysosomale, le PERT a restauré environ 6 % de l'activité de la protéine déficiente, ce qui a permis de guérir presque complètement toutes les manifestations de la maladie.
Les chercheurs ont également testé PERT sur plus de 14 700 mutations pathogènes non sens enregistrées dans des bases de données cliniques, et ont constaté que la technique était capable de lire la grande majorité d'entre elles.
Il est essentiel de noter que les analyses de sécurité approfondies n'ont détecté aucune lecture à travers les codons d'arrêt naturels (qui doivent fonctionner correctement, car ils indiquent quand la fabrication d'une protéine doit prendre fin), ni aucune altération du transcriptome ou du protéome cellulaire, ni aucune édition hors de propos. Les souris traitées n'ont également montré aucun signe de toxicité due au traitement pendant les 15 semaines de suivi.
Quelles implications cela pourrait-il avoir pour les maladies à prions et autres maladies neurodégénératives ?
S'il est important de souligner que très peu de maladies à prions ou autres maladies neurodégénératives sont causées par des mutations sans signification (la plupart étant dues à des mutations avec changement de sens ou à d'autres altérations), ce travail représente une avancée conceptuelle importante pour le domaine de la thérapie génique et les maladies rares en général.
La technologie de « prime editing » utilisée par PERT est extrêmement polyvalente et précise, et pourrait être adaptée à l'avenir pour traiter d'autres types de mutations génétiques. En effet, plusieurs groupes de recherche, dont notre laboratoire, explorent activement l'utilisation du prime editing et d'autres technologies similaires d'édition génétique afin de développer des thérapies ciblant spécifiquement les mutations responsables des maladies à prions familiales.
De plus, le concept de développement de traitements « indépendants de la maladie » pouvant traiter plusieurs patients atteints de pathologies différentes à l'aide d'une seule composition est particulièrement pertinent pour les maladies rares telles que les maladies à protéines prioniques, où le nombre limité de patients rend difficile le développement de traitements spécifiques à chaque mutation.
En résumé
Cette étude représente une avancée majeure dans le domaine de la médecine de précision, car elle démontre qu'il est possible de développer des thérapies géniques qui n'ont pas besoin d'être conçues individuellement pour chaque mutation, mais qui peuvent bénéficier à des patients atteints de plusieurs maladies différentes. Bien qu'il s'agisse encore de recherche préclinique et que des années d'études supplémentaires soient nécessaires avant qu'elle puisse être appliquée à l'homme, la PERT ouvre la voie à un nouveau paradigme thérapeutique dans lequel un seul médicament pourrait transformer le traitement de centaines de maladies génétiques, bénéficiant potentiellement à des centaines de milliers de patients dans le monde entier.
Lien vers article original.