Auteur : Hasier Eraña
Une étude internationale récemment publiée dans la revue scientifique Acta Neuropathologica (vous pouvez accéder à l'article original, en anglais, ici) a analysé en profondeur la maladie de Creutzfeldt-Jakob génétique (MCJ génétique) causée par la mutation E200K, la forme héréditaire la plus fréquente des maladies à prions humaines. Il s'agit de la plus grande étude réalisée à ce jour, avec des données provenant de 177 personnes touchées, ce qui a permis de mieux comprendre pourquoi cette maladie peut se manifester de manière si différente d'une personne à l'autre.
Une même mutation, mais des symptômes très différents
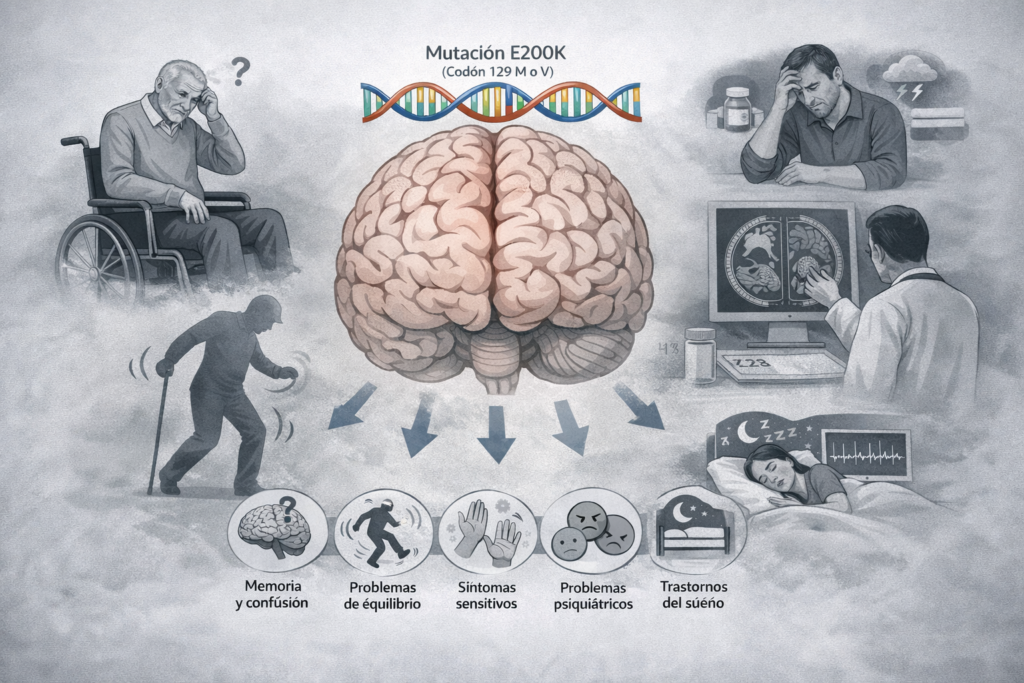
Bien que toutes les personnes incluses dans l'étude partageaient la même mutation génétique (E200K) dans le gène qui produit la protéine prion ou PrP, les chercheurs ont observé une grande variabilité clinique, c'est-à-dire des différences importantes dans : les premiers symptômes, l'âge d'apparition, la durée de la maladie et l'évolution clinique.
La forme la plus fréquente était une détérioration cognitive (problèmes de mémoire, de langage ou de concentration), mais chez de nombreuses personnes, la maladie a commencé autrement : par des problèmes d'équilibre et de coordination, des troubles sensoriels, des symptômes psychiatriques (changements d'humeur, anxiété, comportements étranges) ou, dans certains cas, des troubles du sommeil.
Cela confirme ce que de nombreuses familles savent déjà par expérience : la CEJ génétique ne se manifeste pas de la même manière chez toutes les personnes, même au sein d'une même famille.
Âge de début et durée : différences importantes
L'âge moyen d'apparition de la maladie était d'environ 60 ans, mais avec une grande variation. Certaines personnes sont tombées malades avant 50 ans et d'autres bien après, ce qui renforce l'idée que la mutation ne détermine pas à elle seule le moment où la maladie va se déclarer. Des différences marquées ont également été observées dans la durée de la maladie : dans de nombreux cas, l'évolution a été rapide, de quelques mois seulement. Dans d'autres, en particulier dans certains sous-groupes, la maladie a duré plus d'un an, voire près de deux ans.
Cette étude est donc particulièrement pertinente pour les familles concernées, car, s'agissant du plus grand nombre de cas analysés dans leur ensemble, elle pourrait aider à comprendre pourquoi certaines personnes évoluent très rapidement tandis que d'autres traversent un processus plus long et progressif.
Le rôle du codon 129 : une clé pour comprendre l'évolution de la maladie
L'une des conclusions les plus importantes de l'étude est la confirmation que le codon 129 du gène PRNP (l'acide aminé présent chez chaque individu à la position 129 de la protéine prion ou PrP) influence de manière décisive la manière dont se manifeste et progresse la maladie de Creutzfeldt-Jakob génétique associée à la mutation E200K.
Cette position de la protéine peut présenter deux variantes naturelles, la méthionine (M) ou la valine (V), et chaque personne hérite d'une combinaison des deux. Dans l'étude, ces combinaisons étaient non seulement fréquentes dans des proportions différentes, mais elles étaient aussi clairement associées à des différences cliniques importantes.
Sur les 177 personnes étudiées : 65 % avaient deux copies de méthionine (MM), 23 % avaient une copie de méthionine et une copie de valine (MV) et 5 % avaient deux copies de valine (VV).
L'une des différences les plus marquées entre ces groupes était la durée de la maladie, un aspect particulièrement important pour les patients et leurs familles : les personnes présentant une combinaison MM ont, en moyenne, connu une évolution plus rapide, avec une durée approximative de 4 mois ; les personnes présentant une combinaison MV ont connu l'évolution la plus lente, avec une durée moyenne de 11 mois ; et dans les cas VV, la durée moyenne était intermédiaire, autour de 6 mois.
Ces données confirment que la présence de valine associée à la méthionine (MV) est généralement associée à des évolutions plus longues, tandis que la combinaison MM correspond généralement à des formes plus rapidement progressives.
Différences dans la forme d'apparition et les symptômes prédominants
Bien que la détérioration cognitive ait été le symptôme initial le plus fréquent chez l'ensemble des patients (45 %), l'étude a montré que le type de symptômes peut varier en fonction de la combinaison du codon 129 : Chez les personnes atteintes de MM, le début était plus souvent cognitif, avec des problèmes de mémoire, de langage ou de fonctions exécutives ; tandis que chez les personnes atteintes de MV et VV, les symptômes cérébelleux (difficultés à marcher, instabilité, maladresse) et les symptômes sensitifs étaient relativement plus fréquents.
Parmi l'ensemble des patients analysés, 21 % ont présenté des symptômes cérébelleux, 18 % des troubles sensoriels et 7 % des symptômes psychiatriques ou comportementaux. En outre, l'étude a observé que l'âge d'apparition variait également en fonction du type de symptôme initial : les symptômes cognitifs étaient associés à des âges plus avancés, tandis que d'autres symptômes (tels que les symptômes moteurs) apparaissaient plus tôt.
Sous-types de la maladie : cinq grandes formes cliniques
En combinant les informations cliniques avec d'autres données biologiques, les chercheurs ont identifié cinq grands sous-types de MC génétique E200K, avec une fréquence et une évolution différentes :
- Sous-type classique (MM/MV de type 1) :
Représente environ 67 % des cas.
Il commence généralement par une détérioration cognitive ou des problèmes d'équilibre et évolue rapidement, de manière similaire à la MCJ sporadique. - Sous-type à évolution lente (MM/MV de type 2) :
Environ 7 % des cas.
Il se caractérise par une durée nettement plus longue (jusqu'à 1 à 2 ans) et des tests diagnostiques qui ne sont parfois pas aussi clairs au début. - Sous-type insomnie fatale : Environ 4 %. Elle peut débuter par des troubles du sommeil et une atteinte des régions profondes du cerveau, avec une détérioration cognitive initiale moins importante.
- Sous-types associés à la valine (MV2 et VV2) :
Ils représentent ensemble environ 10 à 11 % des cas.
Ils sont le plus souvent associés à des problèmes de coordination, à des symptômes sensitifs ou psychiatriques, et à une évolution différente de la forme classique.
Tests diagnostiques : ce qui est confirmé et ce qui peut varier
L'étude a également analysé les tests diagnostiques habituels, observant que la plupart des patients présentaient des résultats positifs au RT-QuIC, un test actuellement essentiel pour le diagnostic, et que d'autres tests, tels que la protéine 14-3-3 dans le liquide céphalo-rachidien ou l'électroencéphalogramme, n'étaient pas positifs dans tous les cas, en particulier dans certaines variantes à évolution plus lente ou présentant des symptômes atypiques.
Esto es importante porque explica por qué, en algunas personas, el diagnóstico puede ser más difícil o más tardío, a pesar de tratarse de la misma enfermedad genética.
Pourquoi cette étude est-elle importante pour les familles ?
Ce travail apporte un message clé qui, bien que connu de la communauté scientifique, apporte la confirmation nécessaire que le fait d'avoir la mutation E200K ne signifie pas que la maladie se manifestera d'une seule manière.
De plus, ces résultats confirment que le codon 129 agit comme un modulateur de l'évolution de la maladie, influençant la rapidité de son développement, les symptômes initiaux, la durée totale de la maladie et le type de forme clinique qui se développe chez chaque personne. Cela aide à expliquer pourquoi, même au sein d'une même famille présentant la mutation E200K, les expériences peuvent être très différentes, tant en termes de forme d'apparition que de durée d'évolution.
Ainsi, mieux connaître cette variabilité aide à comprendre les différentes expériences au sein d'une même famille, peut contribuer à réduire le sentiment d'« étrangeté » lorsque la maladie ne suit pas le schéma le plus connu, et apporte des informations précieuses aux médecins et aux équipes cliniques, favorisant une meilleure reconnaissance des différentes formes de la maladie.
De plus, ces connaissances sont essentielles pour progresser vers une médecine plus personnalisée, qui tienne compte non seulement de la mutation, mais aussi d'autres facteurs influençant l'évolution de la maladie.
Un pas de plus vers une meilleure compréhension des maladies à prions
La Fondation espagnole pour les maladies à prions se réjouit de la publication d'études telles que celle-ci, qui élargissent les connaissances scientifiques et, surtout, donnent un nom et une explication à la diversité des expériences vécues par les patients et leurs familles. La recherche ne change pas encore le pronostic de la maladie, mais elle contribue à quelque chose d'essentiel : mieux comprendre ce qui se passe, réduire l'incertitude et jeter les bases de futurs progrès en matière de diagnostic et de traitement.



