Autor: Hasier Eraña
Un estudio internacional recientemente publicado en la revista científica Acta Neuropathologica (puede acceder al artículo original, en inglés, aquí) ha analizado en profundidad la enfermedad de Creutzfeldt-Jakob genética (ECJ genética) causada por la mutación E200K, la forma hereditaria más frecuente de las enfermedades priónicas humanas. Se trata del mayor estudio realizado hasta ahora, con datos de 177 personas afectadas, lo que ha permitido comprender mejor por qué esta enfermedad puede manifestarse de formas tan distintas entre unas personas y otras.
Una misma mutación, pero síntomas muy diferentes
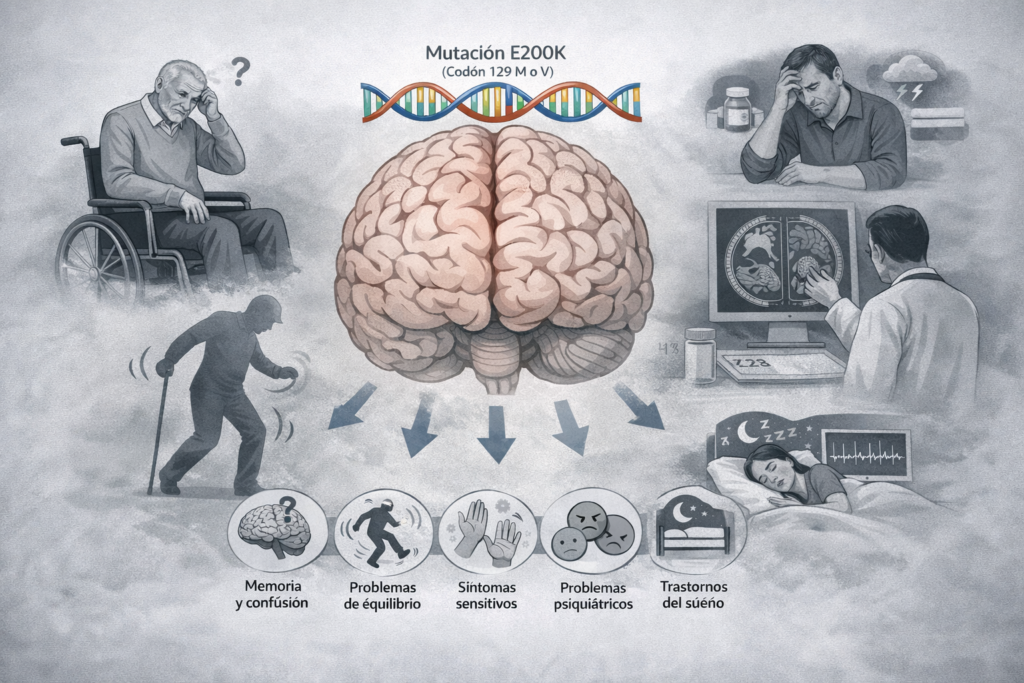
Aunque todas las personas incluidas en el estudio compartían la misma mutación genética (E200K) en el gen que produce la proteína del prion o PrP, los investigadores observaron una gran variabilidad clínica, es decir, diferencias importantes en: los primeros síntomas, la edad de inicio, la duración de la enfermedad y la evolución clínica.
La forma de inicio más frecuente fue el deterioro cognitivo (problemas de memoria, lenguaje o concentración), pero en muchas personas la enfermedad comenzó de otra manera: con problemas de equilibrio y coordinación, alteraciones sensitivas, síntomas psiquiátricos (cambios de ánimo, ansiedad, conductas extrañas) o, en algunos casos, con alteraciones del sueño.
Esto confirma algo que muchas familias ya conocen por experiencia propia: la ECJ genética no empieza igual en todas las personas, ni siquiera dentro de una misma familia.
Edad de inicio y duración: diferencias importantes
La edad media de aparición de la enfermedad fue alrededor de los 60 años, pero con un rango amplio. Algunas personas enfermaron antes de los 50 y otras bastante después, lo que refuerza la idea de que la mutación no determina por sí sola cuándo comenzará la enfermedad. También se observaron diferencias claras en la duración del curso de la enfermedad: en muchos casos, la evolución fue rápida, de solo unos pocos meses. En otros, especialmente en determinados subgrupos, la enfermedad se prolongó más de un año, e incluso hasta casi dos años.
Este estudio es, por tanto, especialmente relevante para las familias afectadas, ya que, al tratarse del mayor número de casos analizados en conjunto, podría ayudar a entender por qué algunas personas tienen una evolución muy rápida mientras que otras atraviesan un proceso más largo y progresivo.
El papel del codón 129: una clave para entender la evolución de la enfermedad
Uno de los hallazgos más relevantes del estudio es la confirmación de que el llamado codón 129 del gen PRNP (el aminoácido que presenta cada persona en la posición 129 de la proteína del prion o PrP) influye de manera decisiva en cómo se manifiesta y progresa la enfermedad de Creutzfeldt-Jakob genética asociada a la mutación E200K.
Esta posición de la proteína puede presentar dos variantes naturales, metionina (M) o valina (V), y cada persona hereda una combinación de ambas. En el estudio, estas combinaciones no solo fueron frecuentes en proporciones distintas, sino que se asociaron claramente a diferencias clínicas importantes.
En el conjunto de 177 personas estudiadas: El 65 % tenía dos copias de metionina (MM), el 23 % tenía una copia de metionina y una de valina (MV) y el 5 % tenía dos copias de valina (VV).
Una de las diferencias más claras entre estos grupos fue la duración de la enfermedad, un aspecto especialmente relevante para pacientes y familias: las personas con combinación MM tuvieron, de media, una evolución más rápida, con una duración aproximada de 4 meses; las personas con combinación MV presentaron la evolución más lenta, con una duración media de 11 meses; y en los casos VV, la duración media fue intermedia, alrededor de 6 meses.
Estos datos confirman que la presencia de valina junto a metionina (MV) se asocia, en general, a cursos más prolongados, mientras que la combinación MM suele corresponder a formas más rápidamente progresivas.
Diferencias en la forma de inicio y los síntomas predominantes
Aunque el deterioro cognitivo fue el síntoma inicial más frecuente en el conjunto de pacientes (45 %), el estudio mostró que el tipo de síntomas puede variar según la combinación del codón 129: En personas con MM, el inicio fue más frecuentemente cognitivo, con problemas de memoria, lenguaje o funciones ejecutivas; mientras que en personas con MV y VV, fueron relativamente más frecuentes los síntomas cerebelosos (dificultades para caminar, inestabilidad, torpeza) y los síntomas sensitivos.
En el total de pacientes analizados el 21 % debutó con síntomas cerebelosos, el 18 % con alteraciones sensitivas y el 7 % con síntomas psiquiátricos o conductuales. Además, el estudio observó que la edad de inicio también variaba según el tipo de síntoma inicial: los síntomas cognitivos se asociaron a edades más avanzadas, mientras que otros síntomas (como los motores) aparecieron antes.
Subtipos de enfermedad: cinco grandes formas clínicas
Combinando la información clínica con otros datos biológicos, los investigadores identificaron cinco grandes subtipos de ECJ genética E200K, con distinta frecuencia y evolución:
- Subtipo clásico (MM/MV tipo 1):
Representa aproximadamente el 67 % de los casos.
Suele comenzar con deterioro cognitivo o problemas de equilibrio y tiene una evolución rápida, similar a la ECJ esporádica. - Subtipo de evolución lenta (MM/MV tipo 2):
Aproximadamente 7 % de los casos.
Se caracteriza por una duración claramente más prolongada (hasta 1–2 años) y pruebas diagnósticas que a veces no son tan claras al inicio. - Subtipo tipo insomnio fatal:
Alrededor del 4 %.
Puede debutar con trastornos del sueño y afectación de regiones profundas del cerebro, con menos deterioro cognitivo inicial. - Subtipos asociados a valina (MV2 y VV2):
En conjunto representan alrededor del 10–11 % de los casos.
Se asocian con mayor frecuencia a problemas de coordinación, síntomas sensitivos o psiquiátricos, y a una evolución distinta de la forma clásica.
Pruebas diagnósticas: qué se confirma y qué puede variar
El estudio también analizó las pruebas diagnósticas habituales, observando que la mayoría de los pacientes presentaron resultados positivos en el RT-QuIC, una prueba clave actualmente para el diagnóstico; y que otras pruebas, como la proteína 14-3-3 en el líquido cefalorraquídeo, o el electroencefalograma, no fueron positivas en todos los casos, especialmente en algunas variantes de evolución más lenta o con síntomas atípicos.
Esto es importante porque explica por qué, en algunas personas, el diagnóstico puede ser más difícil o más tardío, a pesar de tratarse de la misma enfermedad genética.
¿Por qué es importante este estudio para las familias?
Este trabajo aporta un mensaje clave que, aunque conocido por la comunidad científica, aporta la necesaria confirmación de que tener la mutación E200K no significa que la enfermedad vaya a manifestarse de una única manera.
Además, estos resultados confirman que el codón 129 actúa como un modulador del curso de la enfermedad, influyendo en la rapidez de la evolución, los síntomas iniciales, la duración total de la enfermedad y el tipo de forma clínica que desarrolla cada persona. Esto ayuda a explicar por qué, incluso dentro de una misma familia con la mutación E200K, las experiencias pueden ser muy diferentes, tanto en la forma de inicio como en el tiempo de evolución.
Así, conocer mejor esta variabilidad, ayuda a comprender experiencias diferentes dentro de una misma familia, puede contribuir a reducir la sensación de “rareza” cuando la enfermedad no sigue el patrón más conocido, y aporta información valiosa para médicos y equipos clínicos, favoreciendo un mejor reconocimiento de las distintas formas de la enfermedad
Además, este conocimiento es fundamental para avanzar hacia una medicina más personalizada, que tenga en cuenta no solo la mutación, sino también otros factores que influyen en la evolución de la enfermedad.
Un paso más hacia una mejor comprensión de las enfermedades priónicas
Desde la Fundación Española de Enfermedades Priónicas celebramos la publicación de estudios como este, que amplían el conocimiento científico y, sobre todo, ponen nombre y explicación a la diversidad de vivencias de pacientes y familias. La investigación no cambia todavía el pronóstico de la enfermedad, pero sí contribuye a algo esencial: entender mejor lo que ocurre, reducir la incertidumbre y sentar las bases para futuros avances en diagnóstico y tratamiento.



