Author: Hasier Eraña
An international study recently published in the scientific journal Acta Neuropathologica (you can access the original article, in English, here) has conducted an in-depth analysis of genetic Creutzfeldt-Jakob disease (genetic CJD) caused by the E200K mutation, the most common hereditary form of human prion diseases. This is the largest study conducted to date, with data from 177 affected individuals, which has provided a better understanding of why this disease can manifest itself in such different ways from one person to another.
The same mutation, but very different symptoms
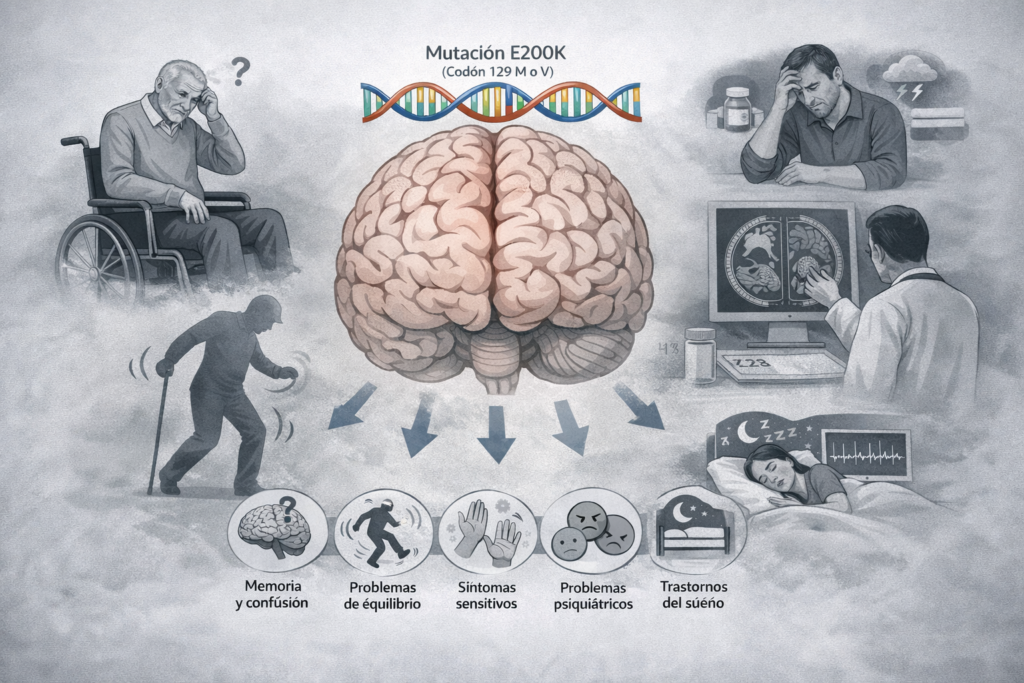
Although all individuals included in the study shared the same genetic mutation (E200K) in the gene that produces the prion protein or PrP, researchers observed significant clinical variability, i.e., important differences in: initial symptoms, age of onset, duration of the disease, and clinical progression.
The most common onset was cognitive impairment (problems with memory, language or concentration), but in many people the disease began in other ways: with problems with balance and coordination, sensory disturbances, psychiatric symptoms (mood swings, anxiety, strange behaviour) or, in some cases, sleep disturbances.
This confirms something that many families already know from personal experience: genetic ECJ does not start the same way in everyone, even within the same family.
Age of onset and duration: important differences
The average age of onset of the disease was around 60 years, but with a wide range. Some people became ill before the age of 50 and others well after, reinforcing the idea that the mutation alone does not determine when the disease will begin. Clear differences were also observed in the duration of the disease course: in many cases, the progression was rapid, lasting only a few months. In others, especially in certain subgroups, the disease lasted more than a year, and even up to almost two years.
This study is therefore particularly relevant for affected families, as it covers the largest number of cases analysed together and could help to understand why some people experience very rapid progression while others undergo a longer, more gradual process.
The role of codon 129: a key to understanding the evolution of the disease
One of the most significant findings of the study is the confirmation that the so-called codon 129 of the PRNP gene (the amino acid present in every person at position 129 of the prion protein or PrP) has a decisive influence on how genetic Creutzfeldt-Jakob disease associated with the E200K mutation manifests and progresses.
This protein position can have two natural variants, methionine (M) or valine (V), and each person inherits a combination of both. In the study, these combinations were not only frequent in different proportions, but were also clearly associated with important clinical differences.
Of the 177 individuals studied: 65% had two copies of methionine (MM), 23% had one copy of methionine and one of valine (MV), and 5% had two copies of valine (VV).
One of the clearest differences between these groups was the duration of the disease, an aspect that is particularly relevant for patients and families: people with the MM combination had, on average, a more rapid progression, with an approximate duration of 4 months; people with the MV combination had the slowest progression, with an average duration of 11 months; and in VV cases, the average duration was intermediate, around 6 months.
These data confirm that the presence of valine alongside methionine (MV) is generally associated with longer courses, while the MM combination tends to correspond to more rapidly progressive forms.
Differences in onset and predominant symptoms
Although cognitive impairment was the most common initial symptom in the patient group (45%), the study showed that the type of symptoms can vary depending on the combination of codon 129: In people with MM, the onset was most often cognitive, with problems with memory, language, or executive functions; whereas in people with MV and VV, cerebellar symptoms (difficulty walking, instability, clumsiness) and sensory symptoms were relatively more common.
Of the total number of patients analysed, 21% presented with cerebellar symptoms, 18% with sensory disturbances and 7% with psychiatric or behavioural symptoms. In addition, the study found that the age of onset also varied according to the type of initial symptom: cognitive symptoms were associated with older ages, while other symptoms (such as motor symptoms) appeared earlier.
Subtypes of disease: five major clinical forms
By combining clinical information with other biological data, researchers identified five major subtypes of genetic CJD E200K, with varying frequency and progression:
- Classic subtype (MM/MV type 1):
Accounts for approximately 67% of cases.
It usually begins with cognitive impairment or balance problems and progresses rapidly, similar to sporadic CJD. - Slow-progressing subtype (MM/MV type 2):
Approximately 7% of cases.
It is characterised by a significantly longer duration (up to 1–2 years) and diagnostic tests that are sometimes unclear at the outset. - Fatal insomnia subtype: Approximately 4%. It may begin with sleep disorders and affect deep regions of the brain, with less initial cognitive impairment.
- Valine-associated subtypes (MV2 and VV2):
Together, they account for around 10–11% of cases.
They are most commonly associated with coordination problems, sensory or psychiatric symptoms, and a different progression from the classic form.
Diagnostic tests: what is confirmed and what may vary
The study also analysed standard diagnostic tests, noting that most patients tested positive on RT-QuIC, currently a key diagnostic test, and that other tests, such as 14-3-3 protein in cerebrospinal fluid or electroencephalogram, were not positive in all cases, especially in some slower-progressing variants or those with atypical symptoms.
Esto es importante porque explica por qué, en algunas personas, el diagnóstico puede ser más difícil o más tardío, a pesar de tratarse de la misma enfermedad genética.
Why is this study important for families?
This study conveys a key message which, although known to the scientific community, provides the necessary confirmation that having the E200K mutation does not mean that the disease will manifest itself in a single way.
Furthermore, these results confirm that codon 129 acts as a modulator of the course of the disease, influencing the speed of progression, initial symptoms, total duration of the disease, and the type of clinical form that each person develops. This helps explain why, even within the same family with the E200K mutation, experiences can be very different, both in terms of onset and progression time.
Thus, gaining a better understanding of this variability helps to comprehend different experiences within the same family, can contribute to reducing the feeling of ‘strangeness’ when the disease does not follow the most familiar pattern, and provides valuable information for doctors and clinical teams, promoting better recognition of the different forms of the disease.
Furthermore, this knowledge is essential for advancing towards more personalised medicine, which takes into account not only the mutation, but also other factors that influence the progression of the disease.
One step closer to a better understanding of prion diseases
At the Spanish Prion Disease Foundation, we welcome the publication of studies such as this one, which expand scientific knowledge and, above all, give a name and explanation to the diverse experiences of patients and families. The research does not yet change the prognosis of the disease, but it does contribute to something essential: better understanding what is happening, reducing uncertainty and laying the foundations for future advances in diagnosis and treatment.



